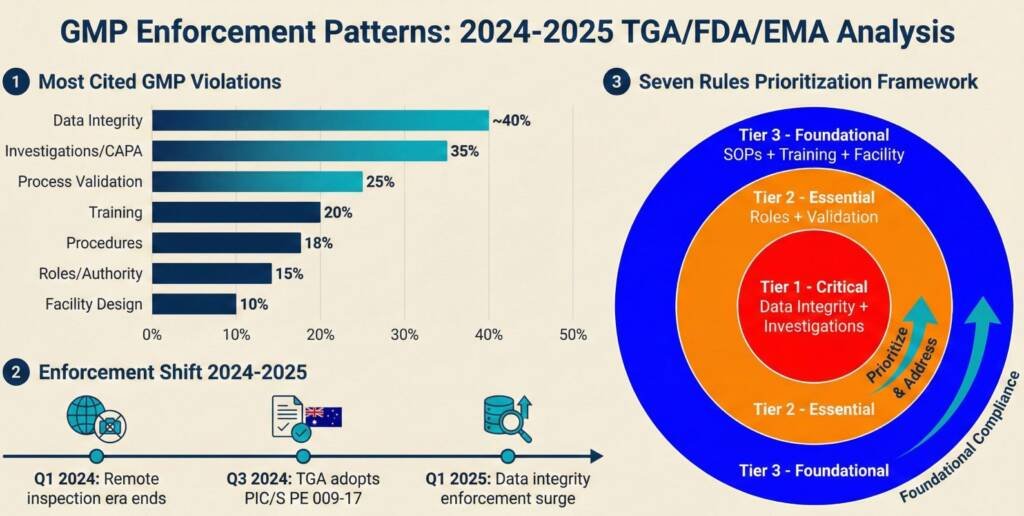
When the regulator arrives for a GMP audit, they assess one underlying question: Can this organisation consistently produce medicine that won’t harm anyone? Recent enforcement data reveal that seven specific GMP requirements account for approximately 70% of significant findings.
These aren’t esoteric technical standards. They’re foundational operational systems that regulators commonly assess every manufacturer to demonstrate from the first commercial batch. These areas reflect recurring inspection focus and enforcement trends, not a guarantee of inspection outcomes or a substitute for a complete pharmaceutical quality system
Australian pharmaceutical manufacturers face acute pressure in 2026. The TGA’s adoption of PIC/S Guide PE 009-17, effective September 2025, aligned Australia’s inspection expectations with international standards. Simultaneously, the regulator shifted from educational to enforcement by litigation, imposing substantial financial penalties. The medicinal cannabis and compounding sectors experienced this most dramatically, with companies receiving fines exceeding $300,000 for GMP non-compliance.
Rule 1: Design Facilities for Contamination Control From Day One
Pharmaceutical facility design must prevent contamination through strategic layout, unidirectional flow, appropriate cleanroom classification, and validated HVAC systems. Remediation of design flaws costs 10-50 times as much as proper initial construction.
The October 2025 Indeus Life Sciences case exemplifies a catastrophic loss of facility control. European inspectors documented “live rodents and birds in storage areas,” “animal faeces on walls,” and “black mould deposits on ceilings above production equipment.” These findings represented fundamental facility envelope breaches and HVAC failures—you cannot procedure your way out of design failures.
Personnel flow control with physical gowning barriers
Cleanroom classification supported by monitoring data
HVAC pressure cascades with alarmed differentials
Surface design with minimal dead spaces
Facility design documentation must be in place before manufacturing begins. When inspectors find hygiene failures, they assess whether facility design made proper control physically possible.
Rule 2: Validate Every Process That Touches Product Quality
Process validation provides documented evidence that manufacturing processes consistently produce products meeting specifications. TGA routinely look for lifecycle validation—process design, qualification, and continued verification—not just initial qualification batches.
The July 2025 Glenmark warning letter illustrates why. FDA cited the company for an inability to control the dissolution process—the Cpk was 0.33, indicating the process couldn’t reliably stay within specification limits. Having validated years ago didn’t excuse ongoing incapability.
Modern lifecycle validation requires:
Stage 1—Process Design: Define Critical Quality Attributes and Critical Process Parameters through development studies and risk assessment.
Stage 3—Continued Process Verification: Statistical process control during routine production, detecting drift before creating out-of-specification batches.
When inspectors see repeated process failures, they question whether the process was ever truly validated. The validation plan, protocols, reports, and ongoing verification data must demonstrate process understanding and control.
Rule 3: Write Procedures That Match Actual Practice
Standard Operating Procedures must accurately describe work performance, be written in unambiguous language, be regularly reviewed, and actually followed during execution. FDA regulation 21 CFR 211.100(b) requires written procedures be followed and documented contemporaneously—”procedure-practice gap” is immediate non-conformance.
Most GMP failures trace to gaps between written procedures and actual practice. Either procedures describe idealised processes nobody follows, or they contain so much detail that operators skip critical steps.
Effective SOPs share characteristics:
Written at the executor competency level
Tested by non-expert staff attempting to follow them
Contain only necessary information.
Include decision points with clear criteria.
Reference but don’t repeat supporting documents.
During facility tours, inspectors observe work and review relevant SOPs simultaneously. Any deviation between procedure and practice becomes a finding. Procedures must specify what records are created during execution, with every critical step having a signature/date indicating who performed it and when.
Trends in GMP enforcement and compliance priorities across TGA, FDA, and EMA inspections from 2024 to 2025.
Rule 4: Map Every Decision to a Named, Qualified Person
A quality unit must have adequate authority and independence to approve or reject components, products, and production processes, as required by 21 CFR 211.22. Recent enforcement shows regulators holding executive management personally responsible when commercial pressure overrides quality decisions.
21 CFR 211.22 appeared in 54 warning letters in 2025, making it the top enforcement priority. Regulators increasingly cite inadequate resourcing of the quality unit as evidence that management is failing to provide the necessary authority.
Critical authority assignments:
Quality Unit Authority cannot be overridden by production or commercial functions.
The Batch Release Authority requires a designated qualified person review before distribution approval.
Deviation Classification specifies roles that determine severity.
Change Control Approval requires quality unit review before validated system changes.
Inspectors verify job descriptions defining roles and authorities, training records proving qualifications, organisational charts showing quality independence from production, and documentation showing quality actually exercised authority to reject non-conforming materials.
Rule 5: Build Data Integrity Into Every Record-Generating System
All records must meet ALCOA+ principles—Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, and Available. TGA and FDA enforcement in 2024-2025 heavily focused on data integrity violations, including shared credentials, missing audit trails, and retrospective data manipulation.
Pharmaceutical quality rests on documented evidence. Inspectors verify compliance by reviewing records that demonstrate processes were followed, specifications were met, and decisions were appropriate.
Recent data integrity enforcement patterns:
Tower Laboratories (December 2025): FDA demanded retrospective review of all invalidated out-of-specification results, citing missing audit trails and shared credentials
Valpharma International (EU, 2025): Statement of Non-Compliance for unauthorised IT system access allowing undetected data manipulation
Glenmark (July 2025): Invalidating out-of-specification dissolution results without adequate investigation—retesting until obtaining passing results
Data integrity in practice requires:
Electronic systems: Complete audit trails logging every data entry, modification, and deletion with user ID and timestamp
Paper systems: Original data in indelible ink, corrections single-line struck through with reason and initials
Hybrid systems: Electronic raw data retained with complete audit trails—paper printouts alone don’t satisfy expectations
Many operations use instruments that capture electronic raw data but generate paper printouts for batch records. Data integrity requires retaining electronic raw data—paper alone is insufficient since unfavourable results can be selectively deleted before printing.
Rule 6: Train Staff to Demonstrated Competency
Personnel must be qualified by education or supervised training before performing quality-affecting operations. TGA expects competency to be demonstrated through practical assessment, not just session attendance—training records without competency verification fail inspection requirements.
Training programs satisfying inspectors include:
Initial GMP training covering pharmaceutical quality principles
Job-specific training on role-relevant procedures
Competency assessment through practical evaluation
Periodic requalification annually or after role changes
Training on deviations/changes when procedures change
Simply attending training doesn’t prove capability. Effective programs include practical assessments where personnel demonstrate correct procedure execution. For analytical chemists, this means performing tests under observation with results compared to established references. For aseptic operators, media-fill validation demonstrates the capability to maintain sterility.
What fails inspections:
Documenting session attendance without verifying understanding
Generic training does not address the specific procedures personnel perform.
Allowing batch record signatures before demonstrating documented competency.
Relying on “years of experience” without periodic requalification
Rule 7: Investigate Thoroughly—It’s Your First Defence
Every out-of-specification result, deviation, or quality failure requires investigation to identify the root cause and implement corrective actions. The most frequently cited GMP violation in 2024-2025 warning letters was failure to thoroughly investigate batch failures per 21 CFR 211.192.
When manufacturers don’t understand why processes fail, they can’t prevent recurrence—leading to repeated failures and patient harm.
Adequate investigations require:
Objective evidence supporting conclusions
Systematic methodology (fishbone diagrams, 5-whys, fault tree analysis)
Investigation depth proportional to failure severity and recurrence
Multiple contributing factors are considered.
The “retraining” CAPA trap: Concluding that operator error caused failure, with “retraining” as the corrective action, rarely satisfies inspectors. They ask: Why did the trained operator err? Was the procedure unclear? Workload excessive? Equipment difficult? True root cause addresses system design, not just individual performance.
Inspectors verify that investigation reports show a logical progression from failure observation through root cause determination to effective corrective action. Evidence that CAPAs were implemented and effectiveness verified. Trending analysis showing whether similar failures recur despite corrections.
This prioritisation reflects implementation sequencing and inspection risk exposure, not the regulatory importance of any GMP requirement, all of which remain mandatory.
Prioritising GMP Improvements With Limited Resources
Most organisations face limited resources to address multiple GMP gaps simultaneously. Prioritise based on enforcement patterns and inspection-readiness impact:
Tier 1—Address Immediately:
Rule 5 (Data Integrity): Foundation for all other evidence
Rule 7 (Investigations): Primary assessment of quality culture
Rule 1 (Facility Design): Limits what procedures can achieve
This sequence recognises that facility design remediation takes months or years, while data integrity systems and investigation quality improve within weeks through policy changes and management commitment.
Conclusion
The seven golden rules aren’t abstract principles—they’re operational requirements with clear evidence expectations. Recent enforcement actions by TGA, FDA, and EMA show that organisations that fail to meet these requirements face warning letters, recalls, import restrictions, suspensions, and criminal prosecution.
Australian manufacturers in 2025 face heightened scrutiny as TGA enforcement evolves from education to litigation. Organisations building inspection-ready systems now—starting with data integrity foundations and investigation quality—position themselves for sustainable compliance and to survive regulatory scrutiny.
GMP compliance isn’t about perfection. It’s about demonstrable control of known risks through documented systems that people follow in practice. When inspectors arrive, the question isn’t whether you have perfect processes—it’s whether you can prove you understand them, control them appropriately, and investigate failures systematically.
The seven rules help organisations assemble credible evidence of process understanding, control, and investigation capability.
Common Questions and Answers
What is the difference between GMP and cGMP?
The “c” in cGMP stands for “current,” emphasizing that manufacturers must use up-to-date systems and technologies that align with the latest regulatory standards, rather than relying on outdated practices.
How do I ensure Data Integrity (ALCOA+)?
Maintain records that are Attributable, Legible, Contemporaneous, Original, and Accurate by implementing strong access controls, enabling electronic audit trails, and preventing unauthorized data changes through technical and procedural safeguards.
What are the requirements for Personnel Training and Qualification?
Training should be kept current and updated when processes, SOPs, or roles change. Competency should be assessed beyond attendance (e.g., practical demonstrations or knowledge checks), and temporary staff or consultants should meet the same role-based training expectations as full-time personnel.
How is “Validation” different from “Qualification”?
Qualification demonstrates that specific equipment or systems (e.g., HVAC) are fit for intended use through IQ/OQ/PQ, while validation provides documented evidence that an end-to-end process consistently produces product meeting predetermined specifications.
What should be included in a Quality Agreement for outsourcing?
Quality Agreements should clearly define responsibilities between the sponsor and CMO for change control, deviation management, investigations/CAPA, batch disposition, documentation, record retention, and audit rights to ensure ongoing compliance and accountability.
How do I handle “Out-of-Specification” (OOS) results?
OOS results require a structured scientific investigation, including hypothesis-driven root cause analysis and documented assessment of laboratory and manufacturing factors. Retesting must be justified and controlled, and batch release decisions must be based on the full investigation, not on a passing retest alone.
What are the expectations for Cleaning Validation?
Cleaning validation should demonstrate that residues and contaminants are consistently removed to prevent cross-contamination. This includes selecting appropriate residues to test for, defining acceptance limits, and establishing worst-case scenarios based on factors like toxicity, solubility, equipment design, and product carryover risk.
How often should a Product Quality Review (PQR) be conducted?
PQRs are typically performed annually, with trending of critical quality data over time. For low-frequency products, organizations should define an approach that still evaluates product and process performance, potentially leveraging multi-year data to support meaningful trend analysis.
What is the correct procedure for Change Control?
Change control should assess potential impact to product quality, validation status, regulatory commitments, and supply continuity before implementation. This includes risk assessment, required testing or requalification, stakeholder approvals, implementation planning, and effectiveness checks after the change is executed.
Reference:
Facts About the Current Good Manufacturing Practice (CGMP) – Link
This article is provided for educational and informational purposes only. It is intended to support general understanding of regulatory concepts and good practice and does not constitute legal, regulatory, or professional advice.
Regulatory requirements, inspection expectations, and system obligations may vary based on jurisdiction, study design, technology, and organisational context. As such, the information presented here should not be relied upon as a substitute for project-specific assessment, validation, or regulatory decision-making.
For guidance tailored to your organisation, systems, or clinical programme, we recommend speaking directly with us or engaging another suitably qualified subject matter expert (SME) to assess your specific needs and risk profile.