


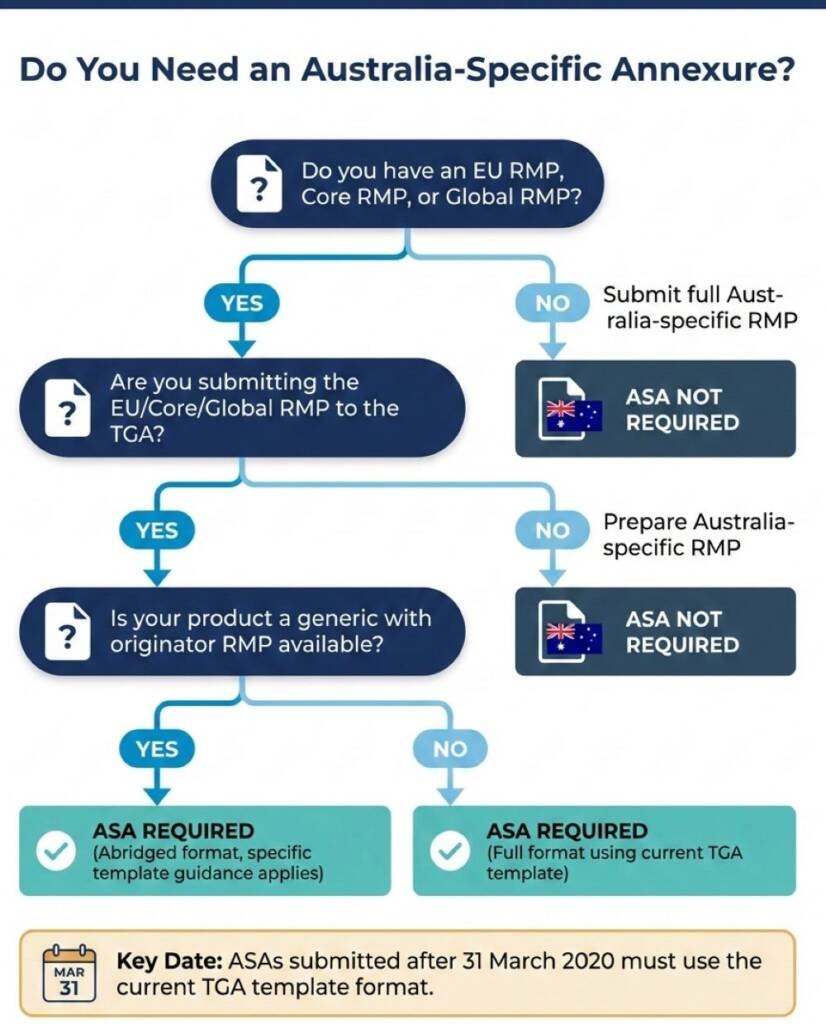
How Should Sponsors Approach the ASA?
The TGA provides a template for the Australian Specific Annexure, and submissions after 31 March 2020 must follow the current format. The template is not simply a checklist—it requires substantive analysis across four domains.
Product overview and registration context. Sponsors document the product’s regulatory history, including approval dates, ARTG numbers, and any previous safety-related variations. This section establishes baseline context for evaluators.
Safety specification alignment. This is where the analytical work happens. Sponsors must compare the summary of safety concerns from the EU RMP against Australian epidemiological data and identify whether differences exist. If Australian disease prevalence differs from Europe, or if risk factors distribute differently across the population, these differences require explicit discussion. The TGA does not expect sponsors to generate new Australian data, but it does expect reasoned analysis of whether existing safety concerns apply equally, less, or more in Australia.
Pharmacovigilance plan localisation. Routine pharmacovigilance activities described in the EU RMP need to be translated for Australian operations. Sponsors explain how adverse event collection, signal detection, and ongoing benefit-risk monitoring will function within Australian systems. For biologicals, additional biovigilance requirements apply.
Risk minimisation measure adaptation. Perhaps the most consequential section. If the EU RMP includes additional risk minimisation measures—educational programmes, controlled access, prescriber restrictions—the ASA must describe how these translate to Australian practice. Sometimes a European measure has no Australian equivalent. Sometimes, Australian healthcare structures enable different approaches. The sponsor’s job is to demonstrate that Australian patients receive equivalent protection through context-appropriate means.
What Does the TGA Actually Evaluate?
When TGA evaluators review Risk Management Plans, they assess whether the documented system adequately addresses the product’s important risks. Their focus is practical: will this RMP, if implemented as written, provide adequate safety monitoring and appropriate risk control for Australian patients?
Evaluators look for evidence of genuine local consideration rather than generic adaptation. Common evaluation concerns include:
- Unchanged safety specifications that fail to address Australian epidemiological differences
- Risk minimisation measures referencing European infrastructure that does not exist in Australia
- Missing justifications for why EU pharmacovigilance activities remain relevant despite different healthcare contexts
- Biovigilance gaps for biological products, where traceability and immunogenicity monitoring requirements differ
The evaluation sits within the broader registration or inclusion process. For prescription medicines, RMP evaluation integrates with clinical and non-clinical assessment at defined milestones. The RMP evaluator’s recommendations feed into the conditions of registration.
The Lifecycle Obligation: RMPs Are Living Documents
Approval of a Risk Management Plan does not end sponsor obligations—it begins the lifecycle management phase. The TGA expects sponsors to maintain their RMPs as living documents that evolve as safety knowledge accumulates.
Mandatory update triggers include:
- New safety concerns identified through signal detection
- Significant changes to risk minimisation measures
- Requests from the TGA based on emerging safety data
- Changes to the EU RMP that affect Australian implementation
- Modifications to Product Information affecting safety messaging
Sponsors should not wait for TGA requests before updating. Proactive maintenance demonstrates good pharmacovigilance practice and avoids situations where RMP documentation lags behind actual safety knowledge.
Common lifecycle maintenance gaps:
✓ Failing to update the ASA when EU RMP revisions occur ✓ Not documenting Australian-specific signal evaluation ✓ Continuing risk minimisation measures past their useful life ✓ Missing records of effectiveness evaluation


