


Why Literature Monitoring Challenges Demand AI Solutions
Manual literature screening is notoriously inefficient. Only a small fraction of search results yield valid ICSRs —often less than 5%. Pharmacovigilance scientists spend most of their screening time reviewing irrelevant publications rather than assessing genuine safety cases.
Teams face interconnected problems: volume overwhelm from thousands of daily publications, low yield despite extensive effort, consistency variability between reviewers, and duplicate complexity across databases. These challenges create concerning dynamics in which overworked teams may narrow search strategies, increasing the risk of missed cases.
AI addresses these challenges by handling high-volume, repetitive filtering—the task humans perform least consistently—while preserving human expertise for medical judgement where it matters most.
Best Practices for Effective Literature Monitoring
Before implementing an AI for monitoring pharmacovigilance literature, organisations must establish robust governance foundations. Technology amplifies existing processes; it cannot compensate for inadequate SOPs.
Essential Governance Elements:
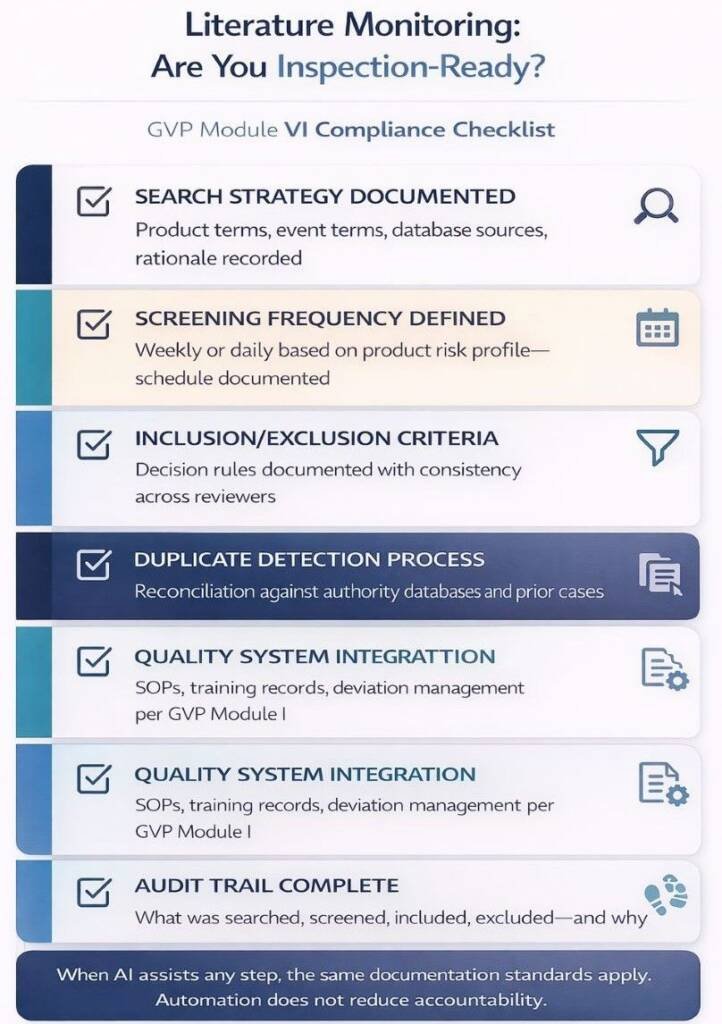
Effective literature monitoring requires documented processes that cover search-strategy definition, frequency scheduling, inclusion/exclusion criteria, duplicate management, and quality system integration. These elements align with GVP Module I quality system expectations.
- Current SOPs document the manual screening process
- Performance baseline exists (sensitivity, specificity, time)
- Audit trail requirements are defined.
- The human oversight model is documented.
- Change control procedures cover model updates.
- Training materials exist for AI-assisted workflows.
The Current Role of Pharmacovigilance Literature Monitoring AI
AI applications in literature monitoring have matured significantly. Current implementations span several complementary functions requiring distinct validation approaches.
- Automated Relevance Classification: AI models categorise abstracts as case-relevant or irrelevant. A 2024 evaluation of large language models achieved 97% sensitivity with 93% reproducibility. The 67% specificity means approximately one-third of flagged articles require human review—an acceptable trade-off compared to manually reviewing everything.
- Named Entity Recognition: Sophisticated systems identify specific ICSR elements: identifiable patient, reporter, suspect product, and adverse event, extracting structured data from unstructured narrative.
- MedDRA Coding Support: AI suggests appropriate adverse event codes. The WHODrug Koda system achieved 89% automation with 97% accuracy on 4.8 million entries.
- Hybrid Architectures: Modern implementations combine rules, retrieval-augmented generation (RAG), and LLM ensembles—improving controllability and reducing hallucination risk.
Human-in-the-Loop: Why PV Expertise Remains Essential
Regulators universally reject fully autonomous decision-making on safety. AI augments human expertise; it does not replace accountability.
CIOMS Human Oversight Models:
- Human-in-the-Loop (HITL): Humans make final decisions with AI providing recommendations. Mandatory for causality assessment, seriousness determination, and reportability decisions.
- Human-on-the-Loop (HOTL): AI executes decisions autonomously while humans monitor aggregate performance. Appropriate for relevance screening and initial filtering.
- Human-in-Command (HIC): Humans set parameters and monitor system performance. Reserved for low-risk applications like duplicate flagging.

For literature monitoring, a typical implementation combines HOTL for initial AI screening with HITL for ICSR confirmation and medical assessment—preserving efficiency while maintaining human accountability.
Future Trends: The Evolution of Literature Surveillance
Pharmacovigilance literature monitoring AI continues evolving toward broader data integration. AI-enabled surveillance is expanding beyond journals into real-world evidence sources: electronic health records, registries, and social media patient reports.
The EMA/HMA workplan (2023-2028) commits to developing AI guidance specific to pharmacovigilance. The EU AI Act classifies many pharmacovigilance applications as high-risk, triggering mandatory transparency and human oversight requirements.
Expect RAG-constrained PV copilots validated against internal SOPs rather than open-ended generative systems. Continuous drift monitoring will become a standard procedure. Bias testing will become an explicit validation requirement.
Conclusion: Balancing Innovation and Regulatory Responsibility
Pharmacovigilance literature monitoring AI delivers genuine efficiency improvements when implemented with appropriate governance. The evidence base—97% sensitivity, 40-50% workload reduction, earlier signal detection—establishes that these systems meet or exceed human performance for well-defined screening tasks.
However, regulatory convergence across the FDA, EMA, CIOMS, and the EU AI Act establishes non-negotiable requirements: risk-based validation, mandatory human oversight, transparent model development, continuous monitoring, and comprehensive documentation.

Key Takeaways:
- AI-assisted literature monitoring is regulatory-acceptable when properly validated.
- Human oversight remains mandatory—AI augments rather than replaces judgment.
- Validation must demonstrate sensitivity ≥95% with confidence intervals.
- Audit trails must reconstruct every screening decision.
- Continuous drift monitoring is now expected, not optional.
Organisations viewing pharmacovigilance literature monitoring AI as a productivity enhancement for expert professionals will successfully navigate validation while capturing efficiency benefits.

