


Your team runs validated systems. Your quality management is documented. Your training records are current. But could you demonstrate that your AI-assisted deviation review remains under regulatory control?
Could you explain to an auditor exactly how your predictive maintenance algorithm influences batch release decisions? This question captures the central challenge of Pharma 4.0 compliance—where digital capability must advance in lockstep with governance maturity.
Pharma 4.0 represents the pharmaceutical industry’s adaptation of Industry 4.0 principles: cyber-physical systems, IoT connectivity, and data analytics applied within GxP-regulated environments. Coined by ISPE in 2017, this framework emphasises that successful implementation depends less on technology selection and more on synchronising digital maturity across validation, culture, and data governance. Organisations treating Pharma 4.0 compliance as a technology project rather than a compliance evolution consistently underestimate the regulatory expectations they face.
What Pharma 4.0 Compliance Means for Quality Teams
The ISPE operating model structures Pharma 4.0 around four foundational pillars that must advance together: Resources, Information Systems, Organisation and Processes, and Culture. Understanding these pillars helps quality leaders identify where their organisation’s digital maturity is actually constrained.
- Resources encompass more than manufacturing equipment. It includes smart facilities with autonomous data acquisition, connected instruments streaming real-time parameters, and digital infrastructure supporting decision-making. Sensors embedded throughout production lines continuously monitor temperature, pressure, humidity, and flow rates—creating the data foundation for advanced analytics.
- Information Systems break down traditional data silos. Quality data flows in real-time rather than sitting in disconnected spreadsheets or legacy tools. This integration supports the traceability that regulators expect and enables the cross-functional visibility that proactive quality management requires.
- Organisation and Processes shift from reactive, manual workflows toward predictive, self-organising systems. Deviation management becomes proactive. CAPA cycles accelerate through automated routing and real-time evidence retrieval. Process controls respond dynamically to incoming data rather than waiting for human intervention.
- Culture remains the most critical yet underestimated dimension. Research consistently indicates that organisational culture presents the greatest challenge to digital transformation. A digital-first mindset requires trust in automated systems and adaptive behaviour from staff at every level. Without cultural readiness, even excellent technology investments fail to deliver their potential.
The Six-Stage Maturity Model That Shapes Your Roadmap
Pharma 4.0 compliance progresses through six distinct stages of digital maturity. Understanding your current position prevents overreach and guides realistic planning. Each stage builds capabilities that enable the next—skipping stages creates governance gaps that surface during regulatory scrutiny.
Most organisations stall at Stage 3 (Visibility). They can monitor processes in real-time but lack the analytical capability to understand causal relationships or predict future outcomes. The transition from “seeing what happened” to “understanding why it happened” requires fundamentally different skills, tools, and governance approaches.
Moving beyond this plateau requires investment in data science capability, governance frameworks for AI-assisted analysis, and cultural willingness to trust algorithmic recommendations—while maintaining the human oversight that regulatory frameworks demand.
What unlocks Stage 5 (Predictability)? Three elements converge: sufficient historical data quality, validated analytical models, and governance structures defining when human review remains mandatory. Without all three, predictive capabilities introduce compliance risk rather than reducing it. Many organisations rushing toward “AI-powered quality” discover that their data foundations cannot support the analytical ambitions.
Validation 4.0—From Reactive Audits to Continuous Verification
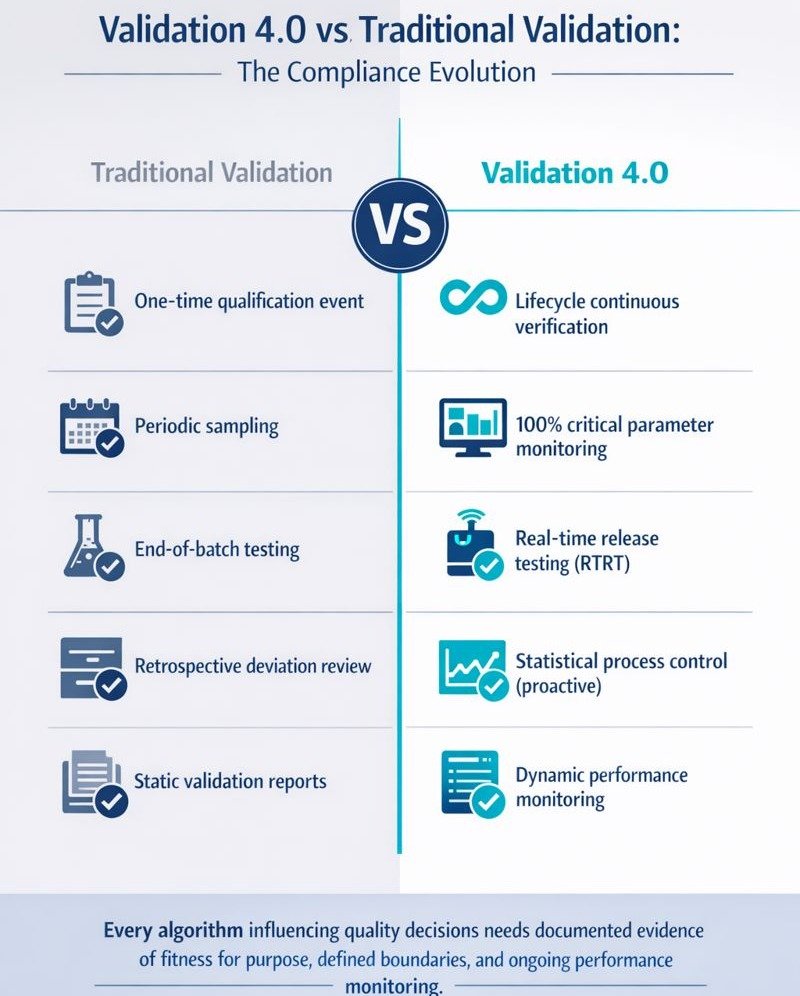
Traditional validation treated system qualification as a one-time event: document requirements, execute protocols, approve the report, and file the evidence. Pharma 4.0 compliance demands a lifecycle approach where validation becomes a continuous process verification.
ISPE’s Validation 4.0 strategy aligns with the FDA’s process validation guidance and advocates three stages: process design, process qualification, and continued process verification. The third stage—often treated as administrative maintenance—becomes the operational centre of Pharma 4.0 compliance.
Continued Process Verification (CPV) under Pharma 4.0 means 100% monitoring of critical parameters rather than periodic sampling. Statistical process control identifies trends before they manifest as deviations. Control charts and capability indices replace end-of-batch testing as primary quality indicators.
Real-time release testing (RTRT) exemplifies this shift. Rather than holding batches for days awaiting laboratory results, in-line sensors and validated models verify Critical Quality Attributes during production. Quality becomes designed into the product, not tested after the fact.
The business case is substantial. Industry case studies show that organisations implementing robust continued process verification (CPV) often report significant reductions in routine analytical workload and measurable decreases in major deviation rates, enabled by earlier trend detection. Benchmarked implementations of real‑time release principles have demonstrated high first‑time‑right performance and major reductions in batch review effort, particularly when supported by review‑by‑exception models. While specific improvements vary by facility and implementation maturity, the overall trend across documented examples is clear: proactive, data‑driven quality consistently outperforms traditional reactive models.
However, continuous verification requires validated models. Every algorithm influencing quality decisions needs documented evidence of fitness for purpose, defined boundaries, and ongoing performance monitoring. This is where Pharma 4.0 compliance intersects directly with AI governance.
Where Regulators Are Heading—QMM, ICH E6(R3), and AI Oversight
Regulatory agencies are not waiting for industry to mature. They are actively architecting frameworks that reward digital capability while increasing scrutiny of those who lag.
FDA’s Quality Management Maturity (QMM) program incentivises practices exceeding baseline CGMP requirements. Facilities demonstrating mature quality systems across five practice areas—management commitment, business continuity, advanced pharmaceutical quality systems, technical excellence, and employee empowerment—may receive regulatory flexibilities, including reduced inspection frequency.
The QMM assessment model progresses from Initial (ad-hoc, reactive) through Managed, Defined, and Quantitatively Managed to Optimising (continuous improvement driven by analytics). Facilities stuck at lower maturity levels face growing disadvantage as the program expands.
ICH E6(R3), finalised in January 2025 and effective in the EU from July 2025, reframes Good Clinical Practice from prescriptive checklists to principles-based, risk-proportionate approaches. Quality by Design and Risk-Based Quality Management shift from optional to foundational. Digital technology integration receives explicit support—eConsent, electronic source data, wearables, and remote monitoring become standard practice when reliability is demonstrated.
AI-driven inspection targeting has arrived. FDA’s June 2025 launch of the “Elsa” system analyses adverse event reports, compliance data anomalies, Form 483 observations, and historical outcomes to prioritise high-risk facilities. Organisations with unresolved quality trends or inadequate CAPA effectiveness will be flagged for focused enforcement. Between July and December 2025, the FDA issued 327 warning letters—a 73% increase over the same period in 2024.
Quality teams must assume that all data visible to regulators—quality metrics, deviation frequencies, CAPA timelines—will be analysed algorithmically. The implications require maintaining audit-readiness continuously rather than episodically.
Starting Without Stalling—Practical First Steps
Pharma 4.0 compliance does not require simultaneous transformation across all domains. Strategic sequencing builds governance capability before scaling AI applications.
Quick wins that build governance muscle:
- Electronic Quality Management System (eQMS) implementation, audit trail establishment, and workflow automation.
- Digital Document Management Systems (DMS) with version control and controlled access.
- Training Management Systems with competency tracking and automated recertification alerts.
- Electronic batch records providing automatic deviation flagging.
These foundational systems create the data infrastructure and governance practices that support advanced applications.

Before piloting AI-assisted processes, establish:
- Data governance framework defining ownership, quality standards, and access controls
- Validation approach for AI/ML systems aligned with GAMP 5 principles
- Human oversight protocols specifying where algorithmic recommendations require expert review
- Change control procedures for model updates and retraining
- Performance monitoring defines acceptable drift thresholds
Assessment checklist for readiness:
- Can you demonstrate data integrity across all digital systems?
- Do you have defined roles for AI governance decisions?
- Is your CPV program capturing 100% of critical parameters?
- Can you explain how any automated decision was reached?
- Would your current documentation satisfy an inspector asking about AI controls?
Cross-functional teams prove essential. Subject matter experts, digital champions, quality assurance, and IT must align before implementation, not during remediation. Executive sponsorship ensures resource allocation survives competing priorities.

