Small pharmaceutical sponsors face an unchanging regulatory reality: TGA pharmacovigilance requirements apply equally to multinationals processing 10,000 cases annually and biotech startups managing 50. The 15-day serious adverse reaction timeline doesn’t account for budget constraints. The 72-hour significant safety issue window doesn’t consider team size.
While large sponsors deploy validated enterprise platforms costing AUD $150,000-500,000 annually, SMEs operate with fractional QPPV support, shared email inboxes, and manual Excel tracking. This creates compliance risk through administrative bottlenecks, causing timeline breaches.
AI pharmacovigilance for small sponsors isn’t about enterprise platforms. It’s building lean workflows in which AI reduces the manual burden by 20-40% while humans retain full decision-making authority.
Your goal:
- Faster administration,
- Unchanged accountability,
- Sustained inspection readiness.
Regulators assess outcomes and oversight, not platform sophistication. TGA inspectors want evidence that you identified, assessed, and reported safety information on time with documented human review and audit trails. They don’t require expensive technology if your lean system produces compliant results.
Why Small Sponsors Face Disproportionate Pharmacovigilance Burden
Three specific pressures distinguish SME pharmacovigilance from enterprise operations:
- Work volume is not correlated with company size. A startup with three ARTG products monitors global literature, responds to spontaneous reports, prepares periodic updates, and maintains inspection documentation regardless of revenue. The work scales to regulatory obligation, not resources.
- Manual processes create timeline jeopardy. The 15-day reporting clock starts when anyone in your organisation receives minimum reportable information—identifiable patient, identifiable reporter, suspect medicine, adverse event. Three days in a shared inbox before QPPV routing consumes 20% of your compliance window.
- Unit economics favour large sponsors. Processing 100 cases at a platform cost of $150,000 equals $1,500 per case. Large sponsors processing 5,000 cases achieve $30 per case. This 50x cost disadvantage forces different architectural choices.
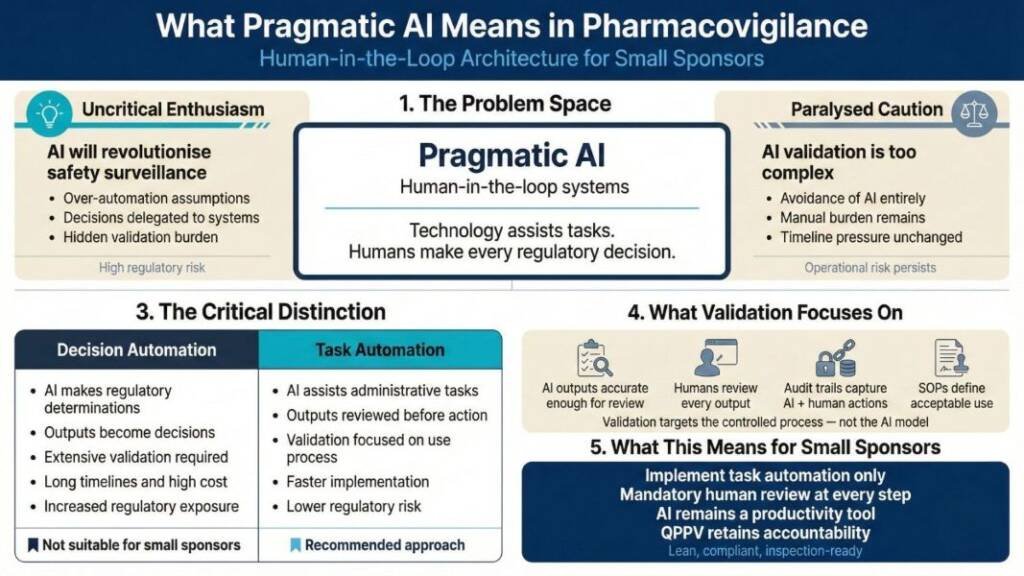
What Pragmatic AI Means: Human-in-the-Loop Architecture
Pharmaceutical AI discussions oscillate between uncritical enthusiasm (“AI will revolutionise safety surveillance”) and paralysed caution (“AI validation is too complex, time-consuming and expensive”). Neither serves small sponsors navigating resource constraints.
Pragmatic AI means human-in-the-loop systems in which technology assists with tasks while humans make every regulatory decision. This architectural choice fundamentally changes validation scope, implementation speed, and regulatory risk.
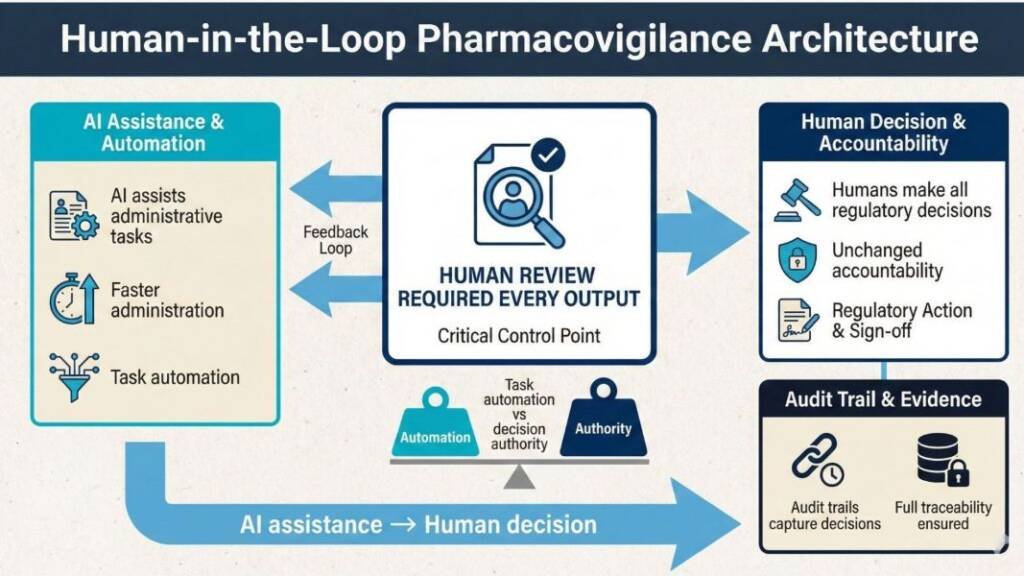
The critical distinction: Are you automating decisions or tasks?
Decision automation requires extensive validation because system outputs directly become regulatory determinations. AI independently classifying event seriousness and triggering TGA reports needs validation proving correct classification across diverse clinical scenarios—expensive, lengthy work requiring substantial documentation and testing.
Task automation reduces the validation burden because humans review every output before action. AI structuring email text into standardised intake fields needs validation to ensure accurate extraction—but human review catches errors before they reach regulatory submissions. The validation focuses on your controlled use process, not the AI’s internal algorithms.
Small sponsors should implement only task automation with mandatory human review. This keeps AI as a productivity tool, rather than a medical device that may require ARTG registration. It also maintains clear professional accountability—the QPPV remains responsible for regulatory decisions, using AI to reduce administrative burden rather than delegate judgment.