


Risk-Based Quality Management Under E6(R3)
E6(R2) introduced risk-based monitoring. E6(R3) expands this to Risk-Based Quality Management (RBQM)—a continuous cycle covering the entire trial lifecycle. This expansion is central to the risk-based quality management requirements of ICH E6.
RBQM under E6(R3) integrates four components:
- Risk identification – What could go wrong? Where are the critical data points?
- Risk control – What processes or checks mitigate those risks?
- Risk communication – How do teams communicate risks across stakeholders?
- Risk review – How frequently do teams revisit assessments?
This is not a one-time exercise. Instead, inspectors expect risk assessments that evolve—teams must update them when safety signals emerge, site performance varies, or protocol amendments change the risk profile.
Common inspection gaps:
- Risk assessments that teams create at study start but never revisit.
- No documented process for risk escalation.
- KRIs and QTLs that teams define but do not actively monitor.
Centralised monitoring plays a larger role under R3. Rather than conducting routine on-site source data verification, sponsors now use statistical monitoring and targeted reviews. As a result, on-site visits become risk-triggered rather than calendar-driven.
Technology, Decentralisation, and Data Integrity
E6(R3) explicitly accommodates wearables, eSource, remote visits, EHR integration, and direct-to-patient models. This legitimises decentralized trials, GCP elements that sponsors already practised but lacked a clear regulatory footing for.
However, technology adoption comes with higher—not lower—data integrity expectations. Specifically, the guideline emphasises complete audit trails, metadata preservation, data flow traceability, and proportionate system validation.
Sponsors running trials with wearables or remote patient-reported outcomes must document how they maintain data integrity across the technology chain—including vendor qualification, validation rationale, and data flow maps.
What you must document:
- Your system validation approach (risk-proportionate).
- Data flow diagrams for each technology component.
- Vendor oversight and qualification evidence.
- ALCOA+ compliance across electronic systems.
Sponsor Oversight Is Sharpened, Not Shared Away
E6(R2) acknowledged that sponsors could delegate trial activities. E6(R3) makes the sponsor oversight ICH E6(R3) expectation explicit: delegation does not transfer accountability.
Sponsors remain responsible for the integrity of delegated work. Therefore, the guideline requires documented oversight plans specifying delegated activities, monitoring methods, escalation pathways, and evidence of review.
This particularly matters for Australian sponsors working with international Service Providers (including CROs). If your Service Provider manages data entry, monitoring, or safety reporting, inspectors will ask for evidence that you maintained active oversight—not that you signed a contract and stepped back.
What inspectors look for:
- Written oversight plans (not just service agreements).
- Vendor performance review records.
- Decision logs showing sponsor engagement on quality issues.
- Clear delegation and responsibility matrices.
Where Do You Start? A Practical Transition Pathway
The transition to E6(R3) does not require rebuilding every system simultaneously. Instead, start with a structured assessment.
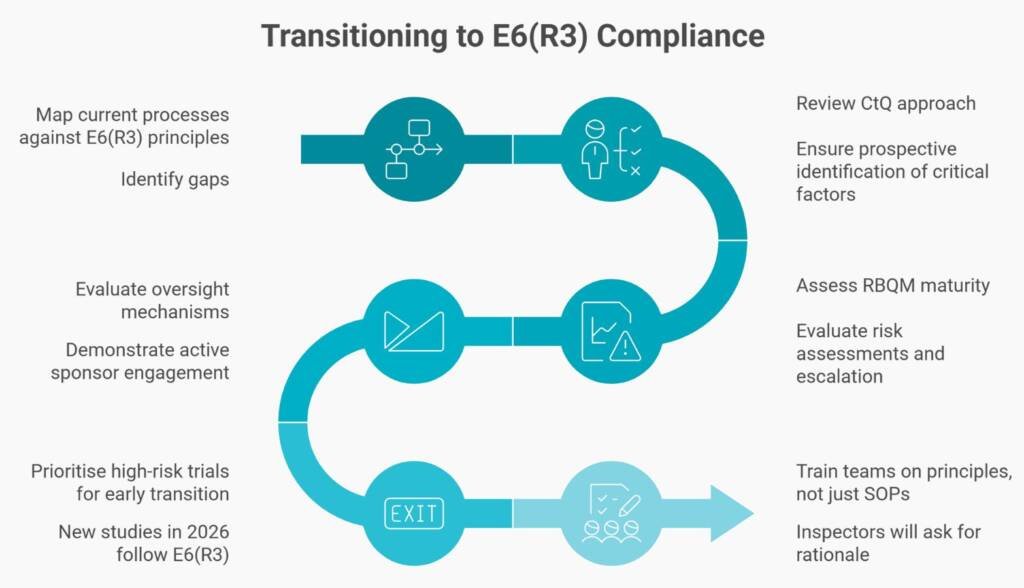
- Step 1: Map your current R2-based processes against the 11 E6(R3) principles. Then identify gaps.
- Step 2: Review your CtQ approach. Do you identify critical factors prospectively with cross-functional input?
- Step 3: Assess your RBQM maturity. Are your risk assessments living documents with defined escalation?
- Step 4: Evaluate your oversight mechanisms. Can you demonstrate active sponsor engagement beyond contract signature?
- Step 5: Prioritise high-risk trials for early transition. New studies starting in 2026 should follow E6(R3) principles from the outset.
- Step 6: Train your teams on principles, not just SOPs. Inspectors will ask staff to explain rationale, not recite procedures.
E6(R3) Readiness Checklist
Use this checklist to assess your current state and prioritise transition:
- You document and maintain a CtQ register for active studies.
- Your team performs risk assessments at study start and reviews them throughout the lifecycle.
- Your RBQM plan aligns with actual trial risks (not a generic template).
- You document oversight plans for CROs and vendors.
- You map data integrity controls (ALCOA+ across electronic systems.
- You document the rationale for the technology validation (proportionate approach).
- You maintain clear delegation and responsibility matrices.
- Your inspection-ready evidence repositories are accessible.
- Staff complete training on E6(R3) principles and rationale.
- Management reviews quality signals and escalation evidence.
Ethics, Participants, and Transparency
E6(R3) reinforces participant-centric language. Informed consent must ensure participants understand not just trial procedures but how teams will collect, use, and protect their data—particularly with digital tools.
Additionally, the guideline strengthens expectations for trial registration and results transparency. For vulnerable populations, sponsors must provide additional safeguards and documented justifications. Patient safety remains the anchor. Every quality system, risk assessment, and oversight mechanism traces back to protecting participants and ensuring data credibility.
What E6(R3) Is Not
To avoid misinterpretation, be clear about what E6(R3) does not permit:
- It is not a relaxation of GCP standards.
- It is not permission for unchecked automation without human oversight.
- It is not a reason to reduce documentation or skip validation.
- It is not an excuse to remove human judgment from safety-critical decisions.
Modernisation means smarter resource application—not fewer controls.
Conclusion: Modernisation With Control
ICH GCP E6(R3) clinical trial modernisation rewards sponsors and sites who design quality into trials from the start, focus oversight where risk is highest, and maintain clear accountability when delegating.
The transition timeline varies by region—EMA compliance is already required, and the TGA has officially adopted the guideline (effective 13 January 2026) with enforcement following the 12-month transition period in 2027. Regardless of jurisdiction, inspectors worldwide will increasingly reference E6(R3) principles.
Those who treat this as an administrative burden will struggle. In contrast, those who treat it as an opportunity to build more defensible, patient-focused trials will find inspections less painful and quality genuinely improved.
The practical next step: Conduct an honest gap assessment against E6(R3) principles. Identify where your processes align, where gaps exist, and what actions will close those gaps before your next inspection.

