


Why Data Management Matters for Australian Clinical Trials
Data management sits at the centre of every credible clinical trial. In Australia, where the Therapeutic Goods Administration (TGA) expects sponsors to demonstrate audit-ready systems, the ability to produce reliable, traceable, and complete trial data directly affects both participant safety and regulatory outcomes.
The TGA adopts ICH GCP guidelines, and with ICH E6(R3) now in effect, Australian sponsors face heightened expectations around data governance, risk-based quality management, and electronic systems validation. Data management is no longer simply about capturing information—it is about demonstrating control throughout the data lifecycle.
This article examines GCP data management from an Australian perspective. It covers the regulatory context, core principles applied in practice, the data lifecycle, expectations for computerised systems, accountability frameworks, and common audit findings. The goal is to help Australian trial teams understand what auditors actually look for and how to build systems that withstand scrutiny.
Australian Regulatory Context for GCP and Data Management
- Australia operates as an ICH-aligned jurisdiction. The TGA recognises ICH E6(R3) as the governing standard for clinical trial conduct, which means Australian sponsors must implement data governance frameworks consistent with international expectations.
- ICH E6(R3) introduced significant changes affecting data management. The guideline now emphasises Quality by Design (QbD), requiring sponsors to identify Critical to Quality (CtQ) factors during protocol development and focus oversight resources accordingly. This shift moves away from blanket 100% Source Data Verification (SDV) toward risk-proportioned monitoring.
- Section 4 of E6(R3) establishes explicit data governance requirements. Sponsors must ensure that computerised systems are validated, that audit trails are reviewed (not merely maintained), and that investigators retain access to their site data regardless of sponsor system hosting. For Australian teams, this means demonstrating active oversight rather than passive documentation.
- During audits, auditors assess whether sponsors have implemented proportionate controls aligned with trial risk. They expect documented evidence of data governance planning, system validation, and ongoing quality monitoring.
Core GCP Data Management Principles Applied in Practice
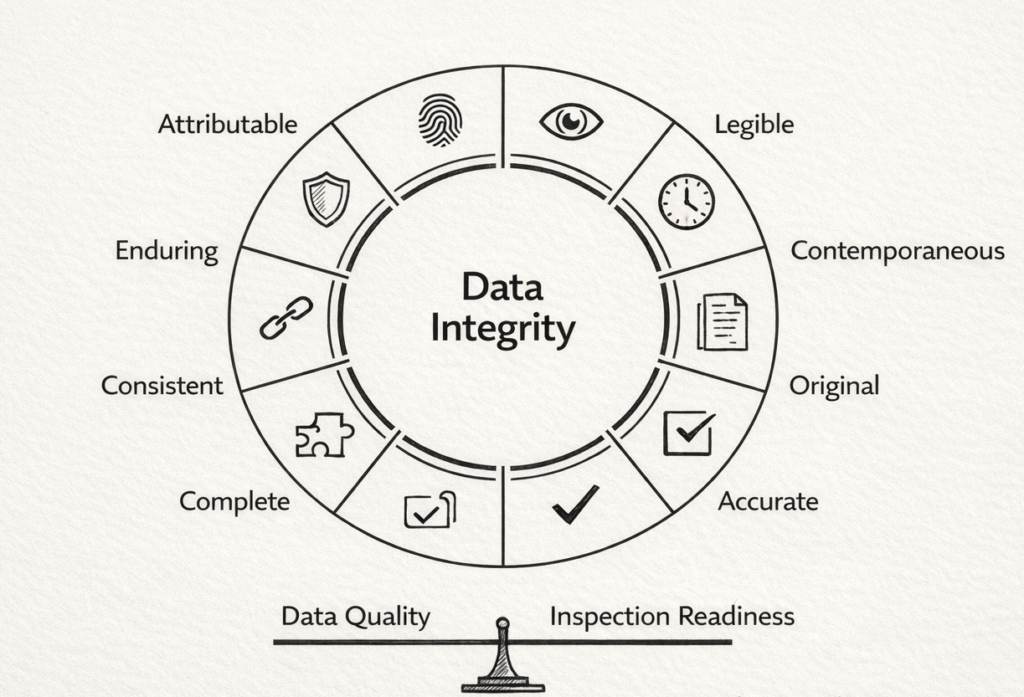
Data integrity remains the foundational requirement. The ALCOA+ framework provides the standard against which auditors evaluate trial data:
| Principle | Practical Application |
|---|---|
Attributable | User authentication links every entry to a specific individual |
Legible | Records remain readable throughout retention periods |
Contemporaneous | Data captured at or near the time of observation |
Original | Source data preserved; certified copies per documented SOPs |
Accurate | Validation checks confirm data reflects reality |
Complete | All required fields populated; audit trails intact |
Consistent | Standard definitions applied across sites and timepoints |
Enduring | Data preserved in accessible, non-proprietary formats |
Available | Retrievable for inspection when required |
Fitness-for-purpose now guides validation efforts. Under E6(R3), Australian sponsors should apply proportionate controls based on the risk each data element poses to participant safety and trial integrity. Primary endpoint data requires more rigorous validation than administrative fields.
Documentation serves as evidence of control. Auditors do not simply want to see that procedures exist—they want evidence that those procedures operate effectively during trial conduct.
The Clinical Data Lifecycle: From Capture to Archiving
Australian trials typically follow a structured data lifecycle with defined controls at each stage.
- Data Capture: Sites enter data into Electronic Data Capture (EDC) systems or paper CRFs. Under E6(R3), investigators bear responsibility for timely and accurate data entry. Edit checks validate data at the point of capture, automatically flagging range violations and logical inconsistencies.
- Data Review and Query Management: Data managers review entries, generate queries for discrepancies, and track resolution. Critically, data managers never change source data directly—only investigators authorise amendments. The query workflow creates an auditable record of every clarification request and response.
- Database Lock: Before statistical analysis, the database undergoes formal lock procedures. All queries must be closed, adverse event reconciliation completed, and investigator sign-offs obtained. The lock report documents completeness verification.
- Archiving: Australian regulations require retention for at least five years post-product approval, though pharmacovigilance data often requires longer retention. Archives must use non-proprietary formats (CSV, XML, PDF/A) to ensure long-term accessibility.
Why Data Management Matters for Australian Clinical Trials
Data management sits at the centre of every credible clinical trial. In Australia, where the Therapeutic Goods Administration (TGA) expects sponsors to demonstrate audit-ready systems, the ability to produce reliable, traceable, and complete trial data directly affects both participant safety and regulatory outcomes.
The TGA adopts ICH GCP guidelines, and with ICH E6(R3) now in effect, Australian sponsors face heightened expectations around data governance, risk-based quality management, and electronic systems validation. Data management is no longer simply about capturing information—it is about demonstrating control throughout the data lifecycle.
This article examines GCP data management from an Australian perspective. It covers the regulatory context, core principles applied in practice, the data lifecycle, expectations for computerised systems, accountability frameworks, and common audit findings. The goal is to help Australian trial teams understand what auditors actually look for and how to build systems that withstand scrutiny.
Australian Regulatory Context for GCP and Data Management
- Australia operates as an ICH-aligned jurisdiction. The TGA recognises ICH E6(R3) as the governing standard for clinical trial conduct, which means Australian sponsors must implement data governance frameworks consistent with international expectations.
- ICH E6(R3) introduced significant changes affecting data management. The guideline now emphasises Quality by Design (QbD), requiring sponsors to identify Critical to Quality (CtQ) factors during protocol development and focus oversight resources accordingly. This shift moves away from blanket 100% Source Data Verification (SDV) toward risk-proportioned monitoring.
- Section 4 of E6(R3) establishes explicit data governance requirements. Sponsors must ensure that computerised systems are validated, that audit trails are reviewed (not merely maintained), and that investigators retain access to their site data regardless of sponsor system hosting. For Australian teams, this means demonstrating active oversight rather than passive documentation.
- During audits, auditors assess whether sponsors have implemented proportionate controls aligned with trial risk. They expect documented evidence of data governance planning, system validation, and ongoing quality monitoring.
Computerised Systems in Australian Clinical Trials
EDC platforms, safety databases, electronic Trial Master Files (eTMF), and laboratory information systems require validation before use in Australian trials.
- Validation Expectations: The TGA expects risk-based validation aligned with GAMP5 principles. High-risk components (randomisation, blinding, audit trails) require comprehensive testing. Lower-risk elements receive proportionate attention. Validation documentation should include requirement specifications, test protocols, and a traceability matrix linking requirements to test results.
- Audit Trail Requirements: ICH E6(R3) mandates that audit trails capture who made changes, when, what was changed, and why. Auditors expect these audit trails to be queryable and reviewed periodically—not simply generated and ignored.
- Common System Issues: Shared login credentials violate the principle of attributability. Inadequate backup procedures create a risk of data loss. Poor change control leads to unvalidated system modifications. Each of these appears regularly in Australian audit findings.
Roles and Responsibilities Across Sponsors, Sites, and Vendors
Clear accountability mapping prevents gaps that auditors identify.
- Sponsors remain responsible for outsourced data management activities. When engaging Contract Research Organisations (CROs), sponsors must maintain oversight through documented vendor qualification, quality agreements, and ongoing performance monitoring.
- Investigators are responsible for data accuracy at their sites. Principal Investigators sign CRF pages confirming that the data reflects the source records. This responsibility cannot be delegated.
- Vendors providing EDC platforms, laboratory services, or safety database hosting must be documented as qualified. E6(R3) requires vendors to report significant non-compliance to sponsors.
A scenario illustrates a common failure: a sponsor engages a CRO for data management. The quality agreement exists but lacks specific key performance indicators. The CRO’s query resolution rate deteriorates. The sponsor lacks monitoring data to detect the problem until a TGA inspection reveals systematic query backlogs. The lesson: oversight requires active metrics, not passive agreements.

Privacy, Data Protection, and Data Residency Considerations
Australian clinical trials must comply with the Privacy Act 1988, which governs the handling of personal information. Cross-border data hosting arrangements—common with cloud-based EDC systems—require documented controls demonstrating adequate protection for participant data transferred overseas.
Participant confidentiality must be balanced with regulatory access rights. Sponsors should ensure consent forms explain that de-identified trial data may be reviewed by regulatory authorities. Technical controls (pseudonymisation, encryption, access restrictions) provide the practical mechanisms to demonstrate privacy compliance.
Quality Management and Risk-Based Data Oversight
E6(R3) requires sponsors to integrate data management into trial-level quality systems. This means identifying Critical to Quality factors during protocol design and monitoring them throughout trial conduct.
- Key Risk Indicators (KRIs) help sponsors detect site-level problems early. Examples include query rates significantly higher than trial average, unusual patterns in adverse event reporting, or data entry concentrated at unexpected times. These indicators should trigger an investigation before problems escalate.
- Quality Tolerance Limits (QTLs) apply at the trial level. If dropout rates exceed protocol assumptions, for instance, the study’s statistical power may be compromised—requiring immediate sponsor action.
Inspection-Ready Checklist
- Data Management Plan approved before first patient enrolled
- EDC system validated with documented test evidence
- Audit trail functionality tested and periodically reviewed
- Query resolution procedures documented in SOPs
- SAE reconciliation completed prior to database lock
- Training records current for all personnel accessing systems
- Backup and disaster recovery procedures tested
Preparing for Sponsor Audits
Auditors assess whether documented procedures translate into actual practice. Australian trial teams should prepare by:
- Aligning SOPs with reality: Review current practice against written procedures. Update documentation or correct practice—but ensure alignment.
- Testing retrieval capabilities: Before audits, verify that archived data, audit trails, and system documentation can be retrieved and presented within reasonable timeframes.
- Briefing key personnel: Ensure staff can explain their roles, describe quality controls, and demonstrate system functionality under questioning.
- Demonstrating oversight evidence: Compile KRI dashboards, quality metrics, and vendor performance summaries that show active monitoring throughout trial conduct.
Conclusion: Strengthening Trust Through Data Discipline
Good clinical practice depends on credible data. Australian trial teams must build data management systems that demonstrate control, not just compliance. Auditors look for evidence that sponsors actively govern data quality throughout the trial lifecycle.
- Practical action steps: Validate systems before use. Review audit trails regularly. Document the data management plan before enrolling participants. Maintain oversight records that demonstrate ongoing monitoring.
Data integrity is essential—it protects participants and produces evidence that regulators and the public can trust. Australian sponsors who commit to disciplined data management establish a defensible, audit-ready foundation for clinical research.
Common Questions and Answers
What data management documentation does the TGA require during clinical trial inspections?
TGA may request Data Management Plans, system validation reports (IQ/OQ/PQ), audit trail exports, query resolution logs, training records, and database lock documentation. These documents verify that documented procedures match actual practice and quality metrics demonstrate ongoing oversight throughout trial conduct.
How does ICH E6(R3) change audit trail requirements for Australian sponsors?
ICH E6(R3) mandates that sponsors actively review audit trails—not merely generate them—with documented procedures specifying review frequency, risk-based scenarios, and corrective actions. This represents a fundamental shift from retrospective documentation to proactive data integrity oversight.
What are ALCOA+ principles and why do TGA inspectors use them?
ALCOA+ defines trustworthy data attributes: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, and Enduring. TGA inspectors use these principles as the benchmark for evaluating data integrity during inspections, with non-compliance in any element potentially compromising trial credibility.
How long must Australian sponsors retain clinical trial data?
Australian regulations require sponsors to retain clinical trial data for minimum 15 years post-completion, with pediatric trials retained until age 25 or 15 years (whichever longer) and gene therapy data retained permanently. Data must remain accessible and readable throughout the entire retention period.
What validation evidence should EDC system documentation include?
Sponsors should maintain User Requirements Specification (URS), validation plan, Installation Qualification (IQ), Operational Qualification (OQ), Performance Qualification (PQ) reports, test protocols with results, change control documentation, and requirements traceability matrices. This documentation demonstrates that systems meet specifications and perform reliably under operational conditions.
What constitutes effective vendor oversight documentation for outsourced data management?
Effective vendor oversight requires documented vendor qualifications, quality agreements specifying responsibilities and performance metrics, evidence of regular monitoring (meeting minutes, KPI reviews), and audit reports with corrective action tracking. This documentation demonstrates that sponsors maintained active supervision throughout the trial rather than delegating-and-forgetting.
What are the most common data management deficiencies identified in TGA inspections?
Common deficiencies include audit trails that exist but are never reviewed, SOPs that don’t reflect actual practice, inadequate training documentation, poor vendor oversight evidence, shared login credentials violating attributability, and incomplete database lock documentation. All 13 TGA inspections conducted in 2023-2024 identified at least one deficiency.
What is Critical to Quality (CtQ) and how does it apply to Australian clinical trials?
Critical to Quality factors are elements directly impacting participant safety, data reliability, or the trial’s ability to answer its scientific question (e.g., primary endpoint data, key safety assessments). ICH E6(R3) requires sponsors to identify CtQ factors early and implement risk-proportioned monitoring focused on protecting these critical elements.
How should Australian sponsors approach database lock documentation and procedures?
Database lock documentation must include the formal request, pre-lock verification evidence, authorization signatures from data management/biostatistics/clinical operations, lock timestamp with audit trail snapshot, and post-lock change procedures. This documentation provides regulatory agencies with confidence that data integrity was maintained throughout analysis and final dataset accuracy.
What does proactive quality management in clinical trials mean under ICH E6(R3)?
Proactive quality management means identifying and preventing data quality issues before they occur through upfront Critical to Quality factor identification, risk assessment, real-time Key Risk Indicator monitoring, and corrective action before issues escalate. This contrasts with retrospective data cleaning and reactive problem-solving approaches from earlier GCP eras.
References
- Therapeutic Goods Administration (TGA) – Good Clinical Practice (GCP) Inspection Program 2023-2024 (Australian Government Department of Health and Aged Care)
- International Council for Harmonisation (ICH) – ICH E6(R3) Guideline for Good Clinical Practice (Final Version, Adopted 6 January 2025)
- National Health and Medical Research Council (NHMRC) – Australian Code for the Responsible Conduct of Research 2018 (NHMRC, Australian Research Council (ARC), Universities Australia, 2018)
- U.S. Food and Drug Administration (FDA) – 21 CFR Part 11 – Electronic Records; Electronic Signatures – Scope and Application (Guidance for Industry, U.S. Department of Health and Human Services, 2003)
- European Medicines Agency (EMA) – EU GMP Annex 11: Computerised Systems (European Commission, Effective 30 June 2011)
- Society for Clinical Data Management (SCDM) – Audit Trail Review: A Key Tool to Ensure Data Integrity – Industry Position Paper (SCDM and eClinical Forum Collaboration, Version PR1, 2021)
- International Society for Pharmaceutical Engineering (ISPE) – GAMP® 5 Guide: A Risk-Based Approach to Compliant GxP Computerized Systems, Second Edition (ISPE, Published August 2022)
- International Council for Harmonisation (ICH) – ICH E8(R1) Guideline: General Considerations for Clinical Studies (Step 5, Adopted 2 November 2021)
Disclaimer
This article is provided for educational and informational purposes only. It is intended to support general understanding of regulatory concepts and good practice and does not constitute legal, regulatory, or professional advice.
Regulatory requirements, inspection expectations, and system obligations may vary based on jurisdiction, study design, technology, and organisational context. As such, the information presented here should not be relied upon as a substitute for project-specific assessment, validation, or regulatory decision-making.
For guidance tailored to your organisation, systems, or clinical programme, we recommend speaking directly with us or engaging another suitably qualified subject matter expert (SME) to assess your specific needs and risk profile.

