


How ICH E6(R3) Changed Everything Your Team Assumed Was Optional
Estimated reading time: 7 minutes
Your sponsor just received an FDA inspection notice. The investigator asks: “Show me validation evidence for every system collecting trial data.” You realise nobody documented User Acceptance Testing for the site’s lab interface, installed six months ago. Under ICH E6(R3), this isn’t an IT oversight—it’s a GCP compliance failure.
Computer System Validation has evolved from procedural IT documentation to a strategic GCP imperative. Consequently, organisations that continue treating CSV as a compliance checkbox rather than a core data governance function risk inspection findings, clinical holds, and the loss of trial data credibility when regulators arrive.
CSV Isn’t IT’s Problem Anymore—It’s Your Inspection Vulnerability
Most teams still treat CSV as something IT validates once and then forgets. A vendor certifies their EDC system. Meanwhile, your CRO confirms their CTMS passed qualification. Sites install lab interfaces without documentation because “the system works fine.” Six months later, an inspector asks for validation evidence and discovers nobody owns the answer.
ICH E6(R3), finalised 6 January 2025 and effective across ICH regions by mid-2025, fundamentally repositioned Computer System Validation within Good Clinical Practice. Specifically, Section 4—an entirely new chapter dedicated to Data Governance—makes clear that CSV isn’t a technical IT exercise delegated to vendors. Rather, it’s a mandatory sponsor accountability directly linked to data integrity, participant safety, and trial reliability.
The guideline defines Computerised Systems Validation as: “A process of establishing and documenting that the specified requirements of a computerised system can be consistently fulfilled from design until decommissioning of the system or transition to a new system.”
This definition carries weight because E6(R3) explicitly requires validation approaches “based on risk assessment considering the intended use of the system and its potential to affect trial participant protection and the reliability of trial results.” Therefore, every system that touches trial data requires documented validation proportional to its criticality.
CSV deficiencies consistently rank among the top 3 inspection findings in EMA, MHRA, and FDA inspections. Everyday observations include inadequate validation documentation, missing audit trails, unvalidated system-to-system interfaces, and a lack of clear accountability for ongoing system performance. Indeed, E6(R3) made an inadequate CSV a formal GCP non-compliance issue. Your systems might work perfectly—but if you can’t prove they’re validated, controlled, and monitored throughout their lifecycle, your trial data lacks the credibility regulators require.
Section 4 Data Governance: The CSV Requirements That Caught Organisations Off-Guard
ICH E6(R3)’s Section 4 represents the most significant evolution in GCP computerised systems requirements in three decades. Furthermore, the guideline establishes non-negotiable requirements that sponsors, investigators, and service providers must address:
System Validation Documentation: Complete evidence from functional requirements through testing, installation, performance verification, change control, and eventual decommissioning.
Risk-Based Validation Approaches: The validation scope must be proportionate to the system’s criticality and impact. Importantly, risk-based doesn’t mean “low-risk systems skip validation”—it means that the validation depth matches the consequences of failure.
Audit Trails and Metadata: Comprehensive audit trails with clear justification for data changes and complete traceability. Every data modification must be attributable, timestamped, and preserved.
User Authentication and Access Controls: Documented user roles and permissions aligned with investigator delegation. When staff leave or roles change, you must update system access to reflect those changes, with documented evidence.
Data Security and Disaster Recovery: Before they’re needed, you must document and test procedures for data backup, recovery, cybersecurity, and business continuity.
Change Control Procedures: Teams must document every upgrade, patch, configuration change, or enhancement through change control to prove validation wasn’t compromised.
Periodic Review Requirements: Perhaps the most significant addition—E6(R3) explicitly states: “Periodic review may be appropriate to ensure that computerised systems remain validated throughout the life cycle of the system.” Thus, validation isn’t a one-time event at system go-live—it’s an ongoing assurance process.
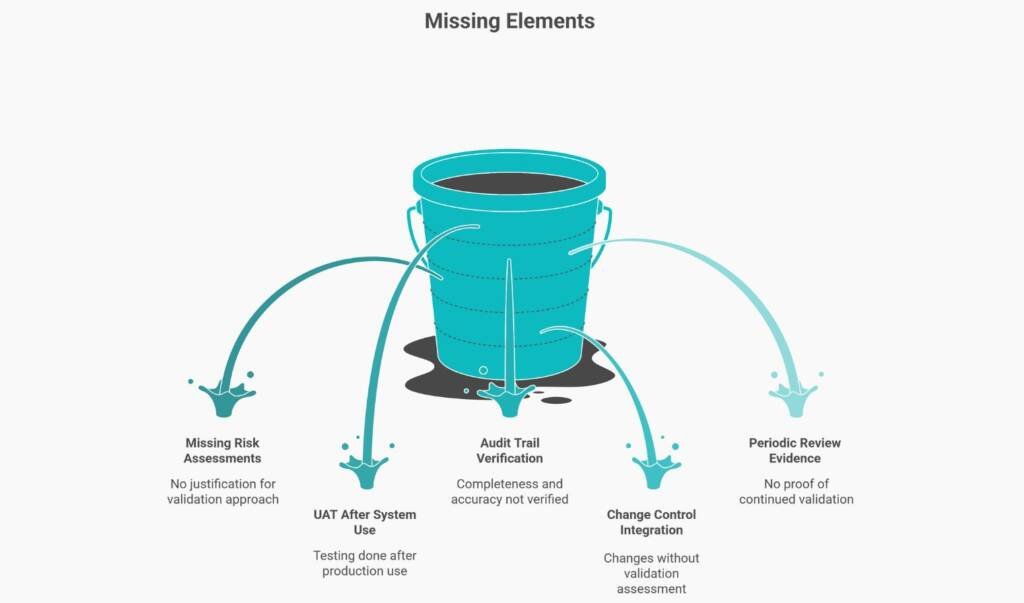
CSV Program Fails When These Five Elements Are Missing
Organisations have SOPs stating “systems will be validated,” but lack operational controls to prove that validation actually occurs. Notably, inspectors identify these gaps within hours of starting document review.
Gap 1: Risk-Based Validation Scope Without Documented Risk Assessments
Teams claim they apply risk-based validation but can’t produce the risk assessments that justified their approach. When asked why System A received full validation but System B only needed vendor qualification, there is no documented decision rationale.
Gap 2: Documented UAT Before Actual System Use
User Acceptance Testing proves the system works in your operational environment before you depend on it for trial decisions. Yet many organisations conduct UAT after systems are already in production. If your UAT report is dated after your first trial data entered the system, you’ve demonstrated you used an unvalidated system—a critical finding.
Gap 3: Audit Trail Completeness Verification
Systems generate audit trails—but does anyone verify they’re complete, accurate, and tamper-proof? Having audit trail functionality enabled isn’t the same as proving it works correctly and remains uncompromised.
Gap 4: Change Control Integration with CSV Lifecycle
Every system changes—through version upgrades, security patches, and configuration modifications. Additionally, each change carries the potential to affect the validation status. When inspectors find that teams implemented system changes without a validation assessment, they’ve identified a system that may be operating in an unvalidated state.
Gap 5: Periodic Review Evidence Proving Systems Remain Validated
E6(R3) introduced explicit periodic review requirements because systems don’t remain validated forever without verification. Organisations that can’t produce periodic review records demonstrate they trusted systems without verification—exactly what validation is designed to prevent.
How to Apply Risk-Proportionate CSV That Actually Works
Risk-based validation sounds efficient until teams realise they’re still validating everything because they can’t defend what’s truly low-risk. However, the solution lies in Critical to Quality factors—elements that directly impact participant safety and data reliability.
Start by classifying systems not by vendor labels but by their role in trial integrity:
High-Risk Systems (Full validation required):
- Systems collecting primary efficacy endpoints
- Systems managing randomisation or blinding
- Systems controlling investigational product accountability
- Systems generating safety reports for regulatory submission
Medium-Risk Systems (Streamlined validation appropriate):
- Systems collecting secondary endpoints
- Systems supporting trial management but not direct data collection
- Systems with compensating manual controls reducing impact
Low-Risk Systems (Vendor qualification plus operational verification):
- Systems supporting general business operations
- Systems with no direct impact on trial data or participant safety
- Systems where alternative data sources provide redundancy
The key: document why each classification applies. When an inspector questions your risk classification, your documented risk assessment—referencing specific CtQ factors—provides defensible rationale.
For High-Risk Systems: Full requirements specification, comprehensive IQ/OQ/PQ, formal UAT, annual periodic review.
For Medium-Risk Systems: Functional requirements mapped to vendor specifications, risk-focused testing, UAT covering essential workflows, and biannual operational verification.
For Low-Risk Systems: Vendor qualification assessment, operational verification at go-live, and annual change control review.
Who Validates What When Systems Cross Organisational Boundaries
Sponsors assume that site-deployed systems are the investigator’s responsibility for validation. Meanwhile, investigators assume sponsor-provided systems absolve them of validation oversight. E6(R3) says both are wrong—and both carry inspection liability.
Sponsor-Deployed Systems at Sites
When sponsors provide EDC, IRT, eCOA, or other systems to sites, sponsors retain accountability for validation. However, investigators must verify that systems function correctly in their local environment, report system issues, and maintain oversight proportionate to the importance of delegated activities.
Investigator-Deployed Systems
When sites deploy their own systems—such as local laboratory interfaces, EMR connections, and pharmacy management systems—investigators become responsible for validation. Specifically, E6(R3) states investigators must “ensure that computerised system requirements in Section 4 are addressed proportionate to risks to participants and data importance.”
This requirement catches many sites unprepared. If trial data flows through site systems before reaching sponsor systems, those site systems must be documented for validation.
The CRO Complication
When sponsors delegate trial conduct to CROs, E6(R3) makes clear: delegation doesn’t eliminate sponsor accountability. Therefore, sponsors must verify CRO systems meet validation requirements, conduct vendor qualification assessments, and maintain oversight through audits.
The inspection risk emerges at system interfaces. When you transfer data from the investigator EMR to the CRO EDC to the sponsor safety database, each interface must be validated. Who validated the interface? Who owns interface changes? Who monitors interface failures? Organisations that can’t answer these questions discover that an inspection vulnerability exists where accountability blurs.
The 90-Day CSV Readiness Roadmap for ICH E6(R3) Compliance
Most teams wait for an inspection notice before organising CSV evidence. By then, it’s too late—validation documentation must exist contemporaneously with system use.
Phase 1: System Inventory and Risk Classification (Weeks 1-4)
Create a comprehensive inventory documenting every system touching trial data. For each system record: name, purpose, validation status, vendor, version, risk classification, responsible person, last assessment date, and planned periodic review date.
Phase 2: Gap Remediation Planning (Weeks 5-8)
For each system lacking adequate validation, determine a remediation pathway: full validation, supplementary validation, vendor qualification upgrade, system replacement, or operational controls.
Phase 3: Control Implementation (Weeks 9-12)
Establish change control procedures requiring validation impact assessment, periodic review schedules, training requirements, issue management processes, and vendor management procedures.
Beyond 90 Days: Continuous Validation Maintenance
CSV readiness requires continuous operational discipline: change control reviews validating every system modification, periodic reviews verifying validation remains adequate, training delivery maintaining user competency, audit trail monitoring, and incident management.
Organisations demonstrating this continuous validation discipline prove to inspectors they control their systems—not the other way around.

Conclusion: CSV as Strategic GCP Foundation
Computer System Validation evolved from an IT documentation exercise to a strategic GCP imperative under ICH E6(R3). Moreover, the guideline’s dedicated Data Governance section makes clear: inadequate CSV equals inadequate GCP compliance.
Organisations treating CSV as optional infrastructure are vulnerable to inspection. Working systems aren’t the standard—defensible, validated, audit-trail-proven systems are.
The path forward requires a cultural shift. CSV isn’t IT’s responsibility—it’s every stakeholder’s accountability. Sponsors’ own validation across their trials. Investigators validate their local systems. Similarly, CROs maintain validated infrastructure.
E6(R3) formalised these expectations because too many organisations learned CSV’s importance only during inspections, rather than beforehand. Those implementing robust CSV programs now—inventory, risk-classify, validate gaps, implement lifecycle controls—build the foundation for reliable trial data that regulatory authorities trust.
Your next inspection will ask: “Prove these systems are validated.” The question isn’t whether your systems work. Rather, it’s whether you can defend that they do.
References
Primary Standards
- ICH E6(R3) Good Clinical Practice Guideline (January 2025)
- GAMP 5 Second Edition: A Risk-Based Approach to Compliant GxP Computerised Systems (2022)
- FDA 21 CFR Part 11 – Electronic Records; Electronic Signatures

