

What Does “Embedding AI” Mean in a GxP Environment?
Embedding AI means integrating AI capabilities into existing documentation workflows as a controlled, validated component—not as a standalone tool operating outside quality systems.
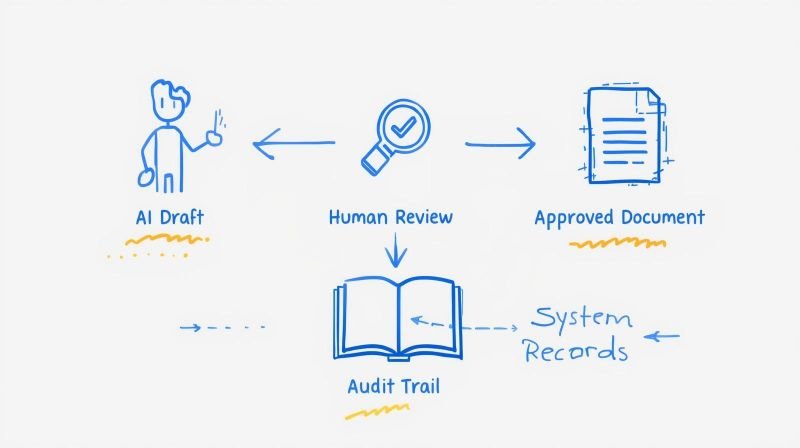
AI functions as a supportive assistant. It drafts content, identifies gaps, and flags documents requiring an update. It does not author final procedures, make release decisions, or replace human judgment on regulatory matters.
This distinction matters because regulators assess AI through the same lens as any GxP process: defined intended use, documented controls, human accountability, and traceable records. AI embedded within your quality system inherits the governance that already governs your documentation practices.
Good Documentation Practice (GDP) principles apply directly. AI outputs require the same review, approval, and version control as manually authored content. The Pharmaceutical Quality System (PQS) provides the governance framework; AI is simply another element within it.
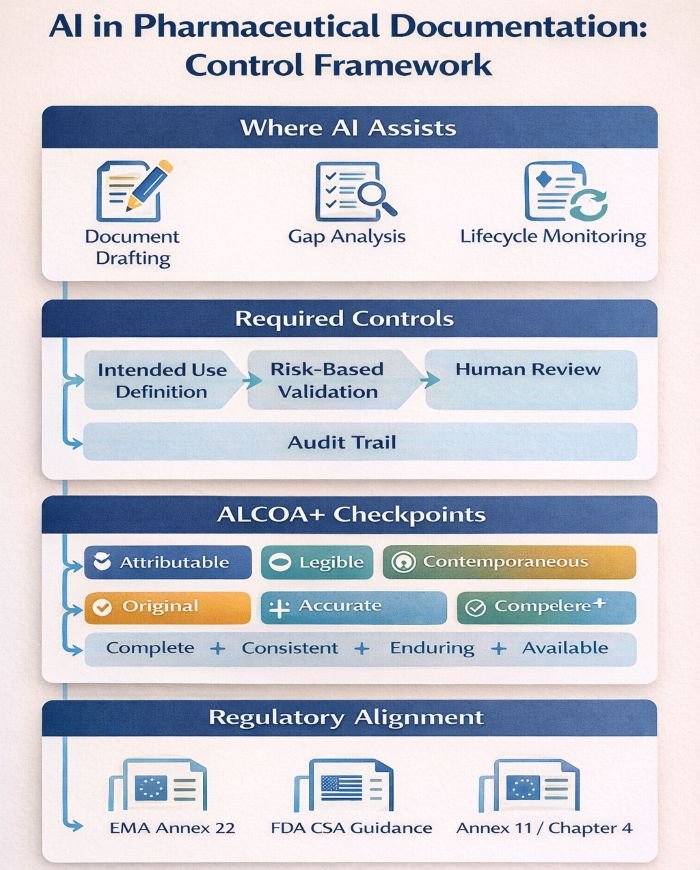
Infographic showing an AI control framework for pharmaceutical documentation with required controls, ALCOA+ checkpoints, and regulatory alignment (EMA Annex 22, FDA CSA, Annex 11/Chapter 4).
AI supports the documentation lifecycle in five high-value ways:
- Drafting & authoring: Generates structured first drafts from approved templates and standard terminology, flags vague phrases, and lets SMEs focus on technical accuracy.
- Regulatory alignment (RAG): Cross-references drafts against authoritative sources (FDA/EMA/ICH and internal policies) to identify gaps and produce traceable gap reports for compliance review.
- Standardisation & structural control: Enforces mandatory sections, consistent terminology, and approved metadata to reduce document inconsistency across sites—an audit pain point.
- Review, training & usability: Creates summaries, checklists, and job aids to improve understanding without changing approved procedures.
- Lifecycle management & change control: Monitors regulatory updates and flags impacted SOPs, pinpointing sections requiring revision to ensure proactive compliance.
What Validation Approach Do AI Systems Require?
- Define intended use. Document precisely what the AI will do within your documentation process. Intended use statements should be specific: “AI tool X generates initial SOP drafts for non-critical procedures, subject to SME review per SOP-123.”
- Demonstrate fit-for-purpose. Annex 22 requires evidence that AI performance meets or exceeds the manual process it supports. This means measuring current baseline performance—error rates, cycle times, consistency—then demonstrating AI outputs achieve equivalent or better results.
- Apply risk-based testing. FDA CSA guidance supports a focused validation effort on high-risk functions. For documentation AI, critical test scenarios include handling complex regulatory requirements, maintaining content accuracy, and ensuring failures are detected during human review.
- Establish change control. AI model updates—whether vendor-supplied or internally developed—require a documented impact assessment. Treat AI providers as critical suppliers, with qualification dossiers that include model version, training data, known limitations, and internal qualification evidence.

AI Documentation Validation Checklist
- Intended use defined and documented
- Baseline manual process performance measured.
- Test scenarios covering critical content requirements
- Human review effectiveness verified.
- Vendor qualification completed (if applicable)
- Change control procedure established for model updates
- Performance monitoring metrics defined.
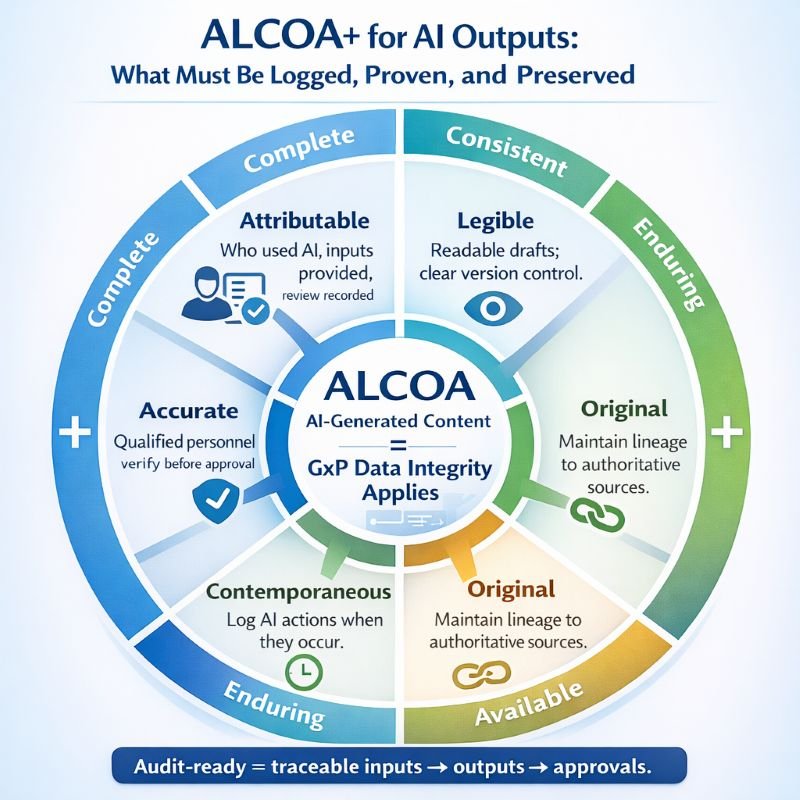
How Do ALCOA+ Principles Apply to AI-Generated Content?
- Attributable requires audit trails that record who initiated AI use, the inputs provided, and how outputs were reviewed. The system must log AI interventions with user identification and timestamps.
- Legible requires AI-generated content to be human-readable and supported by clear version control. Draft outputs must remain distinguishable from approved content during the review process.
- Contemporaneous requires real-time logging of AI interactions. Records must reflect when AI assistance occurred, not when it was later documented.
- Original requires maintaining source data integrity and documenting lineage from authoritative references. RAG systems must identify which sources informed each output.
- Accurate requires verification by qualified personnel before implementation. Human reviewers must confirm content correctness before AI-generated material enters the controlled document system.
- The extended ALCOA+ attributes—Complete, Consistent, Enduring, and Available—ensure comprehensive documentation, standardized processes, long-term archival, and controlled access throughout the AI-assisted documentation lifecycle.
What Governance Framework Supports AI in Documentation?
- Establish AI usage policies and SOPs. Define which AI tools are approved, for what purposes, and under what controls. Policies should specify approval authority, prohibited uses, and documentation requirements for AI-assisted activities.
- Define roles and responsibilities. Clarify who may initiate AI use, who reviews outputs, and who approves final documents. Accountability must be unambiguous; AI should never be listed as the author of record.
- Implement training and competency requirements. Personnel must understand both AI capabilities and limitations. Training should emphasize critical review skills and the essential requirement for professional judgment.
- Complete vendor qualification. Assess AI providers against GAMP 5 principles, with particular attention to model transparency, change notification procedures, and data governance assurances.
What Are the Benefits and Limitations of Embedded AI?
- Benefits. When properly embedded, AI in pharmaceutical documentation delivers measurable improvements, including 30–40% reductions in document revision cycles, 25% fewer procedural errors, and improved inspection readiness. AI identifies inconsistencies that human authors may miss and supports timely updates when regulations change.
- Limitations. Over-reliance remains a risk. AI outputs require the same critical review as manually authored content. Data quality directly affects AI performance; outdated training data produces outdated outputs.
- Explainability gaps may complicate regulatory inspection responses.
- Mitigation. Governance controls such as human review requirements, audit trails, validation evidence, and vendor qualification address these limitations systematically. The goal is controlled augmentation, not autonomous operation.
Future Outlook
Annexe 22’s conservative approach, which restricts generative AI to non-critical applications, reflects current regulatory caution. As the industry demonstrates effective control over AI systems, future guidance revisions may expand permissible applications.
The trajectory is clear: AI will become a permanent, governed component of pharmaceutical quality systems. Organisations that establish robust governance frameworks now will be positioned to adopt expanded capabilities as regulatory confidence matures.
Conclusion
Embedding AI into pharmaceutical documentation can strengthen compliance when properly controlled. AI assists with drafting, gap analysis, standardisation, and lifecycle monitoring. These activities often consume significant QA resources without adding proportionate value.
The regulatory framework now exists. EMA Annex 22, FDA guidance, and established principles from Annex 11 and Chapter 4 define the boundaries. Within those boundaries, AI in pharmaceutical documentation serves as a validated tool that supports human decision-making.

