What Australian Sponsors Must Know in 2026
For Australian sponsors of software-based medical devices, 2026 marks a clear regulatory inflexion point. The TGA’s February 2026 guidance is anchored by its July 2025 report, “Clarifying and Strengthening the Regulation of Medical Device Software, including Artificial Intelligence.” This guidance has materially reset compliance expectations for everyone developing, supplying, or maintaining AI-enabled SaMD (Software as a Medical Device) in Australia. AI medical device software regulation in Australia is now a strategic imperative, not a background activity. This article examines the three pillars driving that reset. It also identifies the risks most sponsors underestimate, and provides a practical compliance checklist.
The Australian Regulatory Framework: Technology-Agnostic but Demanding
Australia’s medical device framework regulates products based on their intended purpose, not the technology they use. Regulation is triggered when software meets the definition of a medical device under section 41BD of the Therapeutic Goods Act 1989 — regardless of whether the underlying system runs on a rule-based algorithm or a large language model.
Technology-agnostic does not mean AI gets a lighter compliance pathway. AI-enabled products introduce novel risks—data drift, algorithmic bias, opaque decision pathways. They must satisfy the same Essential Principles as any other medical device, but need more extensive evidence. The framework is demanding because it makes no concessions for AI novelty. AI medical device software regulation in Australia is grounded in risk. AI introduces risk categories that conventional device regulation was not designed to address directly.

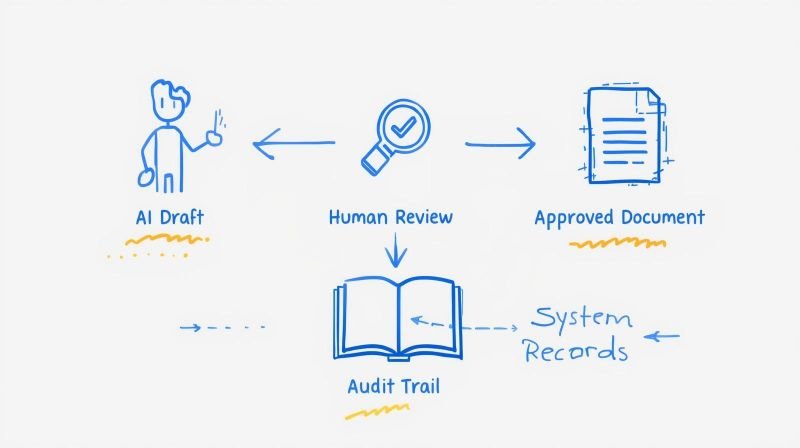
Maintaining a Defensible State of Control Post-Market

International Signals: What EU and US Frameworks Mean for AI Medical Device Software Regulation
Two developments should inform Australian planning. The EU AI Act’s high-risk AI obligations apply from August 2026. All SaMD is automatically classified as high-risk AI, triggering mandatory requirements for explainability, bias detection, and human oversight, in addition to existing MDR obligations. The FDA’s PCCP (Predetermined Change Control Plan) framework, finalised in December 2024, allows manufacturers to pre-define planned device modifications within a single marketing submission. The TGA is examining this approach with a view to international alignment.
No formal Australian PCCP pathway exists yet. Sponsors who structure change control documentation in PCCP-equivalent terms now will be positioned ahead of that transition. Design for the strictest common denominator: build to standards that satisfy TGA, EU, and FDA expectations simultaneously.