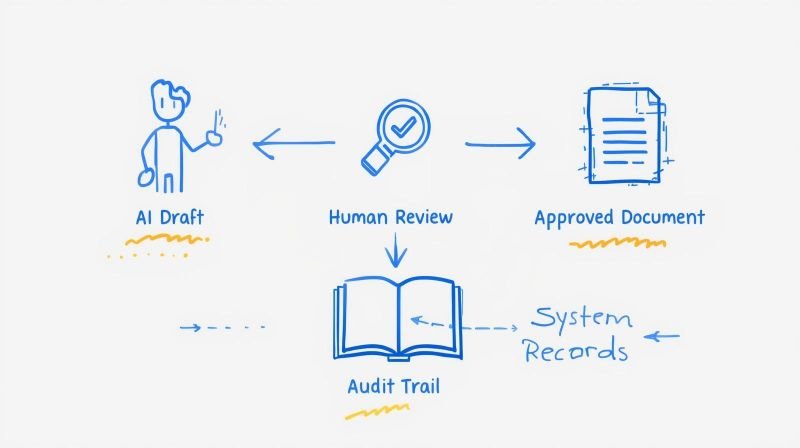
When inspectors request your electronic batch records, clinical trial data, or laboratory results, they check whether your systems meet 21 CFR Part 11 requirements. The regulation defines how pharmaceutical manufacturers, medical device companies, and clinical research organisations demonstrate that electronic records and signatures are trustworthy, reliable, and equal to paper records and signatures.
Since 1997, Part 11 has set data integrity expectations for FDA-regulated industries. Technology has changed, moving from standalone systems to cloud platforms and AI-assisted workflows. Despite these changes, the core principles of the regulation remain the operational standard.
What Is 21 CFR Part 11?
Part 11 defines when electronic records and signatures meet trustworthiness standards. The regulation applies to records created, modified, maintained, archived, retrieved, or transmitted under FDA predicate rules. These rules include core GMP, GCP, and GLP regulations. Predicate rules establish the underlying requirements for maintaining certain records.
Published March 20, 1997, Part 11 granted the FDA regulatory authority to accept electronic records as legally equal to paper records. Organisations must demonstrate they have proper controls over data integrity, system security, and complete audit trails.
When Part 11 Applies
Part 11 applies when predicate rules require records, and you maintain them electronically. Examples include electronic batch production records, LIMS systems recording analytical data, clinical trial EDC platforms, CAPA tracking systems, and pharmacovigilance databases.
FDA’s 2003 guidance distinguishes between required electronic records and voluntary electronic copies. Predicate rules mandate required records when you maintain them electronically. You keep paper as the official record for voluntary copies. Therefore, the whole of Part 11 compliance applies to required electronic records. For voluntary copies, FDA exercises enforcement discretion, provided the paper record remains official and you do not rely on the electronic version for regulatory purposes.
However, hybrid systems add challenges. The key question is which version serves as the official record for inspection. The version that serves this purpose must meet all requirements.
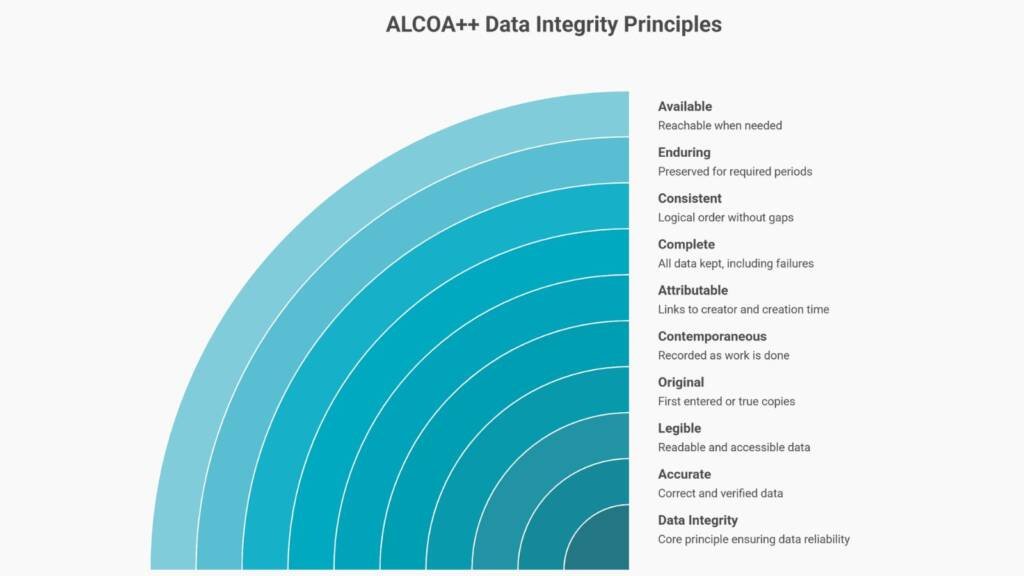
Core Principles: ALCOA+ and Data Integrity
Controls verify that Part 11 records are complete, accurate, secure, and attributable. The FDA expresses these expectations through ALCOA+ principles, which set the operational standard for data integrity.
Recording information contemporaneously—at the time activities occur rather than reconstructing later—is fundamental to meeting these standards. Indeed, FDA warning letters frequently cite violations of this principle.
Furthermore, industry practitioners have extended this to ALCOA++, incorporating additional attributes such as Traceable. While ALCOA++ terminology appears in industry publications, FDA guidance documents don’t formally define it. Nevertheless, the framework reflects evolving best practices for comprehensive data integrity management.
Traceability through audit trails—showing who performed each action, when, and why with sufficient detail to reconstruct events—remains a core requirement under 21 CFR Part 11.10(e). Ultimately, ALCOA++ data integrity principles form the foundation of trustworthy, auditable records in GxP and AI-enabled systems.
Traceable: Audit trails show who did each action, when, and why. There should be enough detail to reconstruct incidents.
Why This Matters
Electronic Records Requirements
System Validation
Match validation scope to the system’s intended use and its effect on product quality and patient safety. Risk-based approaches classify systems into three categories. High-impact systems control key processes, patient data, and regulatory submissions, and have the greatest direct patient impact. In contrast, medium-impact systems support processes with some regulatory or quality consequences. Low-impact systems mainly provide infrastructure and are usually non-GxP.
Specifically, validation typically includes several key stages:
- Installation Qualification (IQ) documents correct installation.
- Operational Qualification (OQ) demonstrates correct performance across operating ranges.
- Performance Qualification (PQ) confirms reliable performance under actual operating conditions.
Auditors look for validation protocols with set acceptance criteria. They want test records showing results, links between requirements and tests, and a documented risk-based rationale for what was tested.
Audit Trails
Additionally, audit trails must operate independently of users; you cannot disable or change them. Retain them for as long as you keep records. Moreover, regularly review audit trails to find unauthorised access or changes.
Common audit trail problems include shared user accounts, trails that can be turned off, missing “reason for change” fields, trails not saved with old records, and a lack of review evidence.
Auditors want to see full audit trails from recent records. They want proof of regular review, such as meeting notes and investigation records. Therefore, procedures should explain who reviews, how often, and when investigations start.
Access Controls
Part 11 requires authority checks, ensuring only authorised individuals use systems, sign records, or perform operations. Implementation includes role-based access control restricting functions to authorised users, approval workflows requiring appropriate authority levels, and administrator privileges separated from normal user access.
In addition, systems must use session timeouts, screen locks requiring re-authentication, logout processes that end sessions, and policies that ban shared passwords.
Auditors look for user access lists showing roles, records of regular access reviews, password policies, and proof that unauthorised actions are stopped.
Data Backup and Retention
Electronic Signatures Requirements
FDA Certification
FDA Enforcement and Common Findings
- Inadequate audit trails: Systems lacking audit trail functionality, trails that can be disabled, or logs not preserved with records
- Shared login credentials: Multiple personnel using the same username/password
- Insufficient validation documentation: Systems deployed without validation or with inadequate testing
- Lack of audit trail review: No procedures or evidence reviews occur
- Inadequate access controls: Personnel having excessive privileges, no periodic review of access rights, or failure to disable terminated employee accounts
Modern Technology Considerations
Cloud Systems and SaaS
AI and Automation
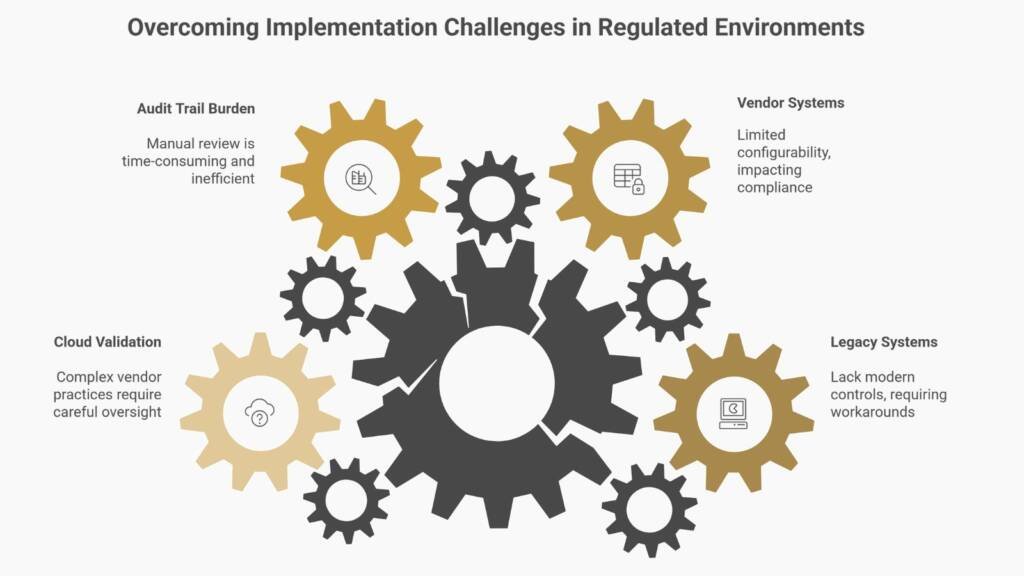
Common Implementation Challenges
Part 11 in 2025 and Beyond
Key Themes for Sustainable Compliance:
- Risk-based thinking: Not every system requires the same level of validation rigor. Focus resources on areas where electronic records have the greatest impact on product quality, patient safety, and regulatory decisions.
- ALCOA+ as operational standard: Data integrity principles provide the conceptual framework. Part 11 provides regulatory specification.
- Continuous compliance: Validation is the starting point. Sustained compliance requires change control, periodic review, audit-trail monitoring, and a quality culture that prioritises data integrity.
- Integration with existing QMS: Part 11 compliance integrates with computerised system validation, change control, training, document management, and all quality system elements.