Introduction
Remember the last time you waited months for a clinical trial to start? Site approvals stalled. Ethics committees requested clarification. The TGA acknowledgement sat in limbo. Your investigational product gathered dust while timelines slipped and budgets strained.
Australia’s clinical trial system doesn’t need to work this way. The Therapeutic Goods Administration (TGA) provides two distinct pathways for supplying unapproved therapeutic goods in clinical trials—the Clinical Trial Notification (CTN) scheme and the Clinical Trial Approval (CTA) scheme. Each serves different purposes, carries different timelines, and requires different levels of TGA involvement before your trial starts recruiting.
For pharmaceutical and biotech sponsors operating in Australia’s regulated healthcare environment, understanding these pathways helps prevent costly delays. This guide explains how the TGA regulates clinical trials, what ethics committees require, when institutional authorisation becomes mandatory, and how to structure your submission so approvals happen in parallel rather than sequence.
Overview: Australia’s Dual Clinical Trial Pathway System
The TGA allows unapproved therapeutic goods to be supplied in clinical trials under two regulatory schemes mandated by the Therapeutic Goods Act 1989 and Therapeutic Goods Regulations 1990. The distinction between these schemes determines how much data the TGA reviews before your trial commences.
The fundamental split:
- CTN scheme (notification) — The TGA doesn’t assess clinical data before the trial starts. The sponsor notifies intent, pays the fee, and may lawfully supply the goods immediately. The ethics committee (called the Human Research Ethics Committee, or HREC, in Australia) is responsible for assessing scientific validity, risk-benefit balance, and ethical acceptability.
- CTA scheme (approval) — The TGA evaluates quality, preclinical, and early clinical safety data before authorising supply. The sponsor submits a formal application with supporting evidence. The TGA conducts a scientific assessment that typically involves multiple evaluation rounds. Only after TGA approval—and ethics committee approval—can the trial commence.
When Each Scheme Applies
The sponsor decides which scheme to use by considering product risk, available data, ethics committee capacity, and institutional requirements. However, CTA is mandatory for specific Class 4 biologicals, regardless of the sponsor’s preference.
Some ethics committees require TGA acknowledgement before beginning their review, even under CTN. Others accept parallel processing. Check institutional policies early—the TGA will accept CTN submissions before ethics approval is finalised, but the trial cannot start until all approvals (TGA notification, ethics approval, institutional authorisation) are documented.

The CTN Scheme: Notification Without Assessment
How It Works
The CTN scheme is a notification process. The sponsor completes an online form through TGA Business Services, pays the application fee ($443 AUD for medicines or biologicals as of 2025), and receives immediate acknowledgement. The trial is deemed notified once the online form is submitted and the fee is paid.
No waiting for TGA review. The sponsor may lawfully supply the investigational product immediately—assuming ethics approval and institutional authorisation are already in place. If you’re notifying before those approvals finalise, you’re responsible for ensuring everything aligns before participants enrol.
The TGA doesn’t evaluate your investigational brochure, preclinical data, or trial protocol at the time of notification. That responsibility sits with the ethics committee, which must assess:
- Scientific validity of trial design
- Balance of risk versus potential benefit
- Overall ethical acceptability
- Adequacy of informed consent processes
When the TGA Can Intervene
Even though CTN doesn’t include an upfront assessment, the TGA retains authority. If the TGA becomes aware that conducting or continuing your trial would be contrary to the public interest—particularly if an unacceptable risk of death, serious illness, or serious injury exists—the TGA can direct the trial not to proceed. At that point, the goods lose their exemption from the Australian Register of Therapeutic Goods (ARTG), and supply becomes unlawful.
The TGA may also request additional information if the trial raises concerns. You’re required to provide details about supply, handling, monitoring, and results if asked.
Note: All these documents can be reviewed during the TGA GCP Inspection.
Documentation Requirements
The online CTN form captures:
- Sponsor details and Australian contact information
- Protocol number and trial description (250-2,500 characters)
- Trial type, therapeutic area, and participant numbers
- Investigational product details: dosage form, route of administration, indication, and GMP license/clearance
- Site details and preceding trial information
- Declaration confirming sponsor responsibility
Critical: If you add new sites to an existing CTN trial, that’s a variation requiring a new fee ($443 AUD per site notification). If you add a new therapeutic good or create a distinct product by changing the notified good, that’s also a fee-triggering variation.
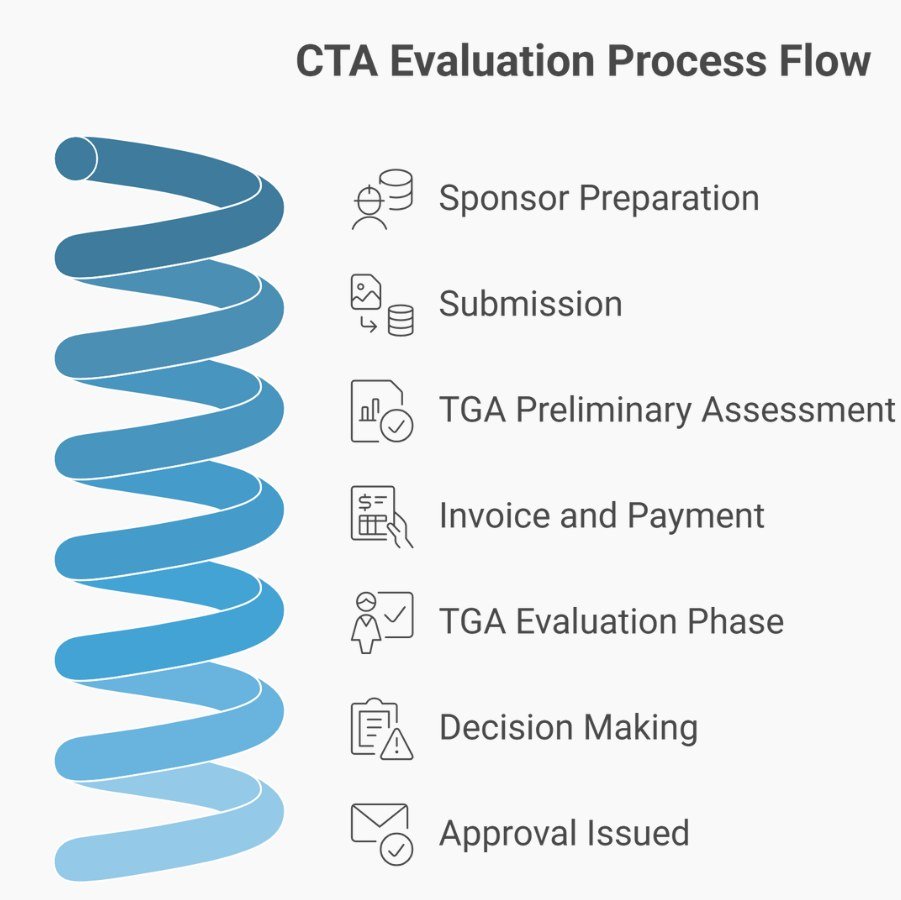
The CTA Scheme: TGA Assessment Before Supply
How It Works
CTA involves a formal application to the TGA for approval to supply unapproved therapeutic goods in a clinical trial. The sponsor submits:
- Part 1: CTA application form with supporting data (quality, preclinical, early clinical safety)
- Part 2: Trial commencement notification (submitted within 28 days of commencing supply at each site)
The TGA conducts a preliminary assessment to ensure sufficient data exists for evaluation. If critical data is missing, the TGA requests it before issuing an invoice. Evaluation begins after payment. The TGA generally completes two rounds of assessment and one round of information requests, though additional rounds may be needed.
Evaluation covers:
- Quality of the therapeutic goods
- Safety of the therapeutic goods
- Compliance with applicable Therapeutic Goods Orders (TGOs)
- Compliance with international guidelines (ICH, FDA, EMA)
- Labelling and traceability
A TGA senior medical officer reviews evaluation reports and recommendations, considering the overall risk-benefit profile. The decision-maker may seek advice from the TGA’s statutory advisory committees. The TGA informs the sponsor whether the trial is approved.
CTA Variations
If you need to change the therapeutic good or any aspect evaluated by the TGA in your original submission, you’ll likely need a variation requiring additional evaluation and a fee payment. Changes predicted to affect product quality, safety, or the trial’s risk-benefit profile trigger assessment. Examples include:
- Significant manufacturing process changes
- Different intended patient group
- Route of administration changes
- Container modifications
Changes not evaluated in the original submission or with no predicted effect on the good or trial safety won’t be assessed by the TGA, but all changes must be communicated to and approved by the ethics committee before implementation.
Fees and Timelines
CTA fees vary by product type and evaluation timeline [2025]:
- Medicines CTA (30-day evaluation): $2,111 AUD
- Medicines CTA (50-day evaluation): $26,240 AUD
- Biologicals CTA: $31,955 AUD
- CTA variations: $580 – $8,718 AUD depending on product and evaluation type
Timeline reality: The TGA doesn’t publish standard review times for CTN or CTA. Contact the TGA for specific timeline inquiries. Most sponsors report CTA taking several months when evaluation rounds and information requests are factored in.